バイオダイレクトメール vol.34 Technical Tips
<どうやってノックダウンしますか?>
一口にRNAiと言っても、RNAiによる遺伝子ノックダウンの方法はいくつもあります。その中でも、下記にあげる化学合成したsiRNAを導入する方法と、siRNA発現ベクターで形質転換する方法は広く用いられています。。手軽に早く結果を得るには化学合成したsiRNAが適しており、長期的なノックダウンの効果を観察するにはsiRNA発現ベクターで形質転換する方が良いといわれています。
- 化学合成したsiRNAを導入する。
- siRNA発現ベクターで形質転換する。
- 酵素を使用してin vitro transcriptionしたsiRNAを導入する。
- 長いdsRNAを導入する。
- DicerやRNase IIIでdsRNAを切断して生じたsiRNAミックスを導入する。
- PCRで合成したsiRNA発現カセットを導入する。
前回のTechnical TipsではRNAiの概略や歴史についてご紹介しましたが、今回はこれらの方法についてそれぞれの概略・メリット・欠点をご紹介します。
1.化学合成したsiRNAを導入する
<概略>
一般的に広く使われている方法です。化学合成した2本のRNA(センス鎖とアンチセンス鎖)をアニーリングしたsiRNAを細胞に導入します。導入されたsiRNAは細胞質でRISCを形成し、ターゲット遺伝子のmRNAの配列特異的な分解を引き起こします。かつては困難だったRNA合成も、合成ケミストリの進歩に伴い、高純度のsiRNAを安定に合成できるようになりました。Dharmacon™社が開発した最新のRNA合成ケミストリ(2’-ACE保護基)は合成速度、収量、品質の点で特に優れており注目されています。

siRNAの模式図
- メリット
- 最も早く結果を得ることができます。
- siRNA合成サービスを利用でき、合成や精製などの手間がかかりません。
- siRNAに各種修飾を施すことができ、蛍光顕微鏡を用いて細胞への導入を確認したり、特殊な修飾でOff-Target効果を抑制したりできます。
- 発現ベクターのような大きな分子を導入する場合に比べて導入が容易です。
- スケールアップが容易です。
- 広く使用されているので、書籍や論文などの情報が充実しています。
- 欠点
- siRNA合成料金が必要です。
- 合成にはある程度の時間が必要です(数日~2週間程度)。
- RNaseによってsiRNAが分解されるため、長期にわたる観察には適しません。
siRNA合成サービスはPCRのプライマーなどに使用されるDNAオリゴの合成サービスに比べて高価であることが多いため、まずは後述の酵素を使用してin vitro
transcriptionしたsiRNAなどの安価な方法でスクリーニングを行い、さまざまな塩基配列の中で最も効果の高かった配列で、siRNAを化学合成し、論文用のデータを取得する、といった方法がとられていたこともありました。しかし、ノックダウン効果を保証したsiRNA設計・合成サービスや、すぐに実験に使えるsiRNA製品が登場し、さまざまな塩基配列のsiRNAを酵素などで合成して試行錯誤するための試薬代や時間を考えると、必ずしも化学合成したsiRNAが高価だとは言えなくなってきています。同時に、そのような新しいサービスや製品を利用することで、設計を行う必要もなくなります。
また、RNaseによる分解を受けにくくする修飾も開発され、以前よりも長期にわたってノックダウン効果を維持することが可能になっています。
2.siRNA発現ベクターで形質転換する
<概略>
この方法も一般的に使われている方法です。たくさんのベクターが開発されていますが、ヘアピン型とタンデム型の2種類に大別できます。ヘアピン型は、プロモーター配列の下流にshRNAの塩基配列を挿入した発現ベクターです。導入された発現ベクターから転写されたshRNAは、核から細胞質へ輸送され、Dicerによるプロセシングを受けてsiRNAと同様の2本鎖siRNAになります。タンデム型はそれぞれがプロモーター配列をもつセンス鎖とアンチセンス鎖の鋳型配列が挿入されたベクターです。それぞれ別々に転写されたセンス鎖とアンチセンス鎖がアニーリングしてsiRNAを形成します。細胞に導入されたプラスミドのコピー数が低い場合には、タンデム型よりもヘアピン型のほうがノックダウン効果が高いという報告があります。
ヘアピン型siRNA発現ベ
クターの模式図
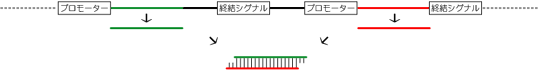
タンデム型siRNA発現ベ
クターの模式図
- メリット
- 細胞内で発現するため、長期間の観察に最適です。
- 抗生物質で導入された細胞を選択(クローン化)することができます。
- 欠点
- 塩基配列を設計する必要があります。
- ベクターに挿入するDNAオリゴが必要です。
- ベクターを構築する手間がかかります。
- ベクターの導入はsiRNAの導入に比べて困難な場合があります。
- 細胞内で発現させたRNAなので修飾することができません。
- 実験系の至適化に時間がかかります。
発現ベクターを使用すれば、安定株をつくることが可能ですが、ベクターの構築後、シークエンシング解析を行い、設計通りのベクターができているかを検証する必要があります。ヘアピン型のsiRNA発現ベクターの場合、シークエンシング解析では細胞内ではヘアピンが確実に形成されるかどうかを明確に確認することはできません。
細胞核内での転写効率や、核から細胞質への輸送、細胞質でのプロセシング(ヘアピン型のsiRNA発現ベクターの場合)など、ベクターを使用する場合にはさまざまなステップが関与します。そのため、siRNAを直接導入する場合に比べて実験結果に影響を及ぼす因子が多くなるため、実験系の至適化にはsiRNAの場合よりも手間がかかります。
3.酵素を使用してin vitro transcriptionしたsiRNAを導入する
<概略>
In vitro transcriptionで使用されるプロモーター(T7、T3、SP6)と組み合わせた鋳型配列からRNA Polymeraseを使用してsiRNAを合成し、精製した後に細胞に導入します。
- メリット
- 安価です。
- siRNA合成サービスに比べてsiRNAを短期間でつくることができます(1-数日)。
- siRNAに修飾を施すことができます。
- 欠点
- 鋳型の設計が必要です。
- 鋳型の準備、合成、精製、アニーリングなどに、手間と技術を要します。
- 酵素で合成するため大幅なスケールアップは困難です。
- RNaseによってsiRNAが分解されるため、長期にわたる観察には適しません。
安価にsiRNAを合成できるため、ノックダウン効果の高い配列を決定するためのスクリーニングに適しています。しかし、ノックダウン効果を保証したsiRNA設計・合成サービスや、すぐに実験に使えるsiRNA製品が登場したため、スクリーニングのために要する手間や試薬代を考えると、必ずしも酵素を使って合成したsiRNAが安上がりであるとは言い切れなくなってきています。
通常、化学合成したsiRNAに比べて、酵素を使用して合成したsiRNAは不完全な長さのsiRNAの割合が高く、Off-Target効果を生じる可能性が高まります。
また、細胞への毒性を考えると精製度の高いsiRNAが必要になります。
4.長いdsRNAを導入する
<概略>
RNAi研究の初期に行われていた方法で、長鎖のdsRNAを導入します。この方法の最大の問題は、哺乳類細胞では使えないことです。長鎖のdsRNAの導入によって、インターフェロン応答が引き起こされ、非特異的に翻訳が阻害された結果、細胞はアポトーシスを起こしてしまうからです。
- 長所
- Dicerによって細胞内で切断され、適切な長さの各種のsiRNAを生じるので、設計が不要です。
- 短所
- 非特異的な細胞応答を引き起こすため、哺乳類の細胞では使えません。
- さまざまな塩基配列のsiRNAが生成されるため、Off-Target効果を生じる可能性が高まります。
5.DicerやRNase IIIでdsRNAを切断して生じたsiRNAのミックスを導入する
長鎖のdsRNAをDicerやRNase IIIで切断した断片(siRNA)のミックスを導入します。さまざまな塩基配列のsiRNAが混ざっていますので、その中のどれが有効だったのかはわかりませんが、ターゲット遺伝子をノックダウンできる確率は高いため、とりあえずノックダウンしてターゲットとなる遺伝子を探索する段階では有用です。
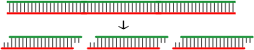
- メリット
- 欠点
- さまざまな塩基配列のsiRNAが導入されるため、Off-Target効果を生じる可能性が高まります。
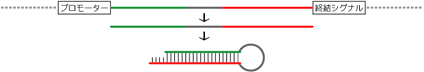
6.PCRで合成したsiRNA発現カセットを導入する
<概略>
比較的新しい方法です。プロモーター - shRNAの鋳型配列 - 転写終結シグナル、という配列で構成されるPCR産物を導入します。
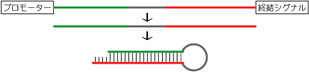
siRNA発現カセットの模式図
- メリット
- 欠点
- 塩基配列を設計する必要があります。
- siRNAよりも導入効率が劣ります。
- PCRを利用するので、大幅なスケールアップは困難です。
ノックダウンの動作機序は、ヘアピン型siRNA発現ベクターと同じです。PCR産物の末端に制限酵素サイトを組み込んでおくことで、ノックダウン効果のあったカセットを利用して容易に発現ベクターを構築することができます。
▼▼Dharmacon™ポータルサイトはこちらから▼▼

