|
|
||||
|
Location:Home > テクニカル情報配信サービス > Pharma Mail |
||||
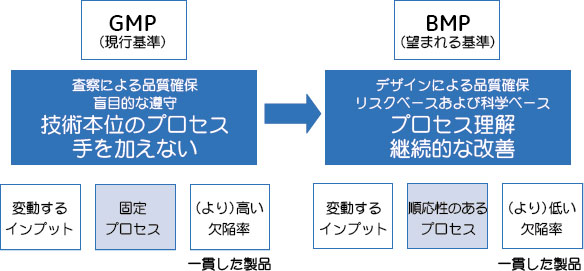
 徹底した査察と広範囲に及ぶバリデーション手順がcGMPの特徴です。プロセスは、初期の段階で「固定」されることが多く、一度申請し、手順を踏んで承認されたら「手をつけない」のが、通常の考え方です。しかし、FDAの新しいPATイニシアティブにより、すべて変わってしまうかもしれません。 下流プロセスにおけるPAT 序説
PAT: Process Analytical Technology(工程分析技術)は、広範な規制イニシアティブ「Pharmaceutical cGMPs for the 21st century - a risk-based approach(21世紀に向けた医薬品cGMP:リスクに基づいたアプローチ)(参考文献1)」の一部として米国FDAが提唱したフレームワークです。FDAはPATを「製造プロセスのモニタリング、分析および管理によってプロセス性能および製品の品質を至適化するためのシステム」と説明しています。PATの全般的な目標は、よりよいプロセス理解と継続的な改善を通じて、常に高品質な製品を製造できるよう保証することにあります。 欠陥率と品質コストの低減今日、徹底してGMPおよびバリデーションに対して努力を重ねているのにもかかわらず、医薬品業界ではいまだに欠陥率が比較的高く(バイオ医薬品全体では1%、特に細胞培養製品では4~5%と推定)、品質確保に多大な支出を強いられています(おそらく、総売上原価[COGS]の25~30%と高い割合)。 PATによって、この状況がよい方向へ変化するという期待が、規制当局および製造業者の取組みの動機となっています。10億分の1(ppb)という低レベルの半導体産業の域に達するのは現実的ではありませんが、改善の余地は確かにあり、今日のCOGSに占める品質コストを削減することも可能なはずです。 GMPに代わるBMPFDAが提唱するように、PATの核心はGMPからBMP(better manufacturing practice、よりよい製造基準)への移行を促すことにあります(図1)。製品の品質を保証するのはリスク評価と科学に基づくデザインです。プロセスは、不変的というよりむしろ順応性があること、また継続的に改善することが新しい基準となります。ここでは、プロセス理解が中心的な役割を担います。FDAがPATのフレームワークに関するガイダンス文書(参考文献2)で述べているように、「本フレームワークは、プロセス理解を基礎とし、業界および当局による新機軸ならびにリスクベースの規制判断を促す」ものです。
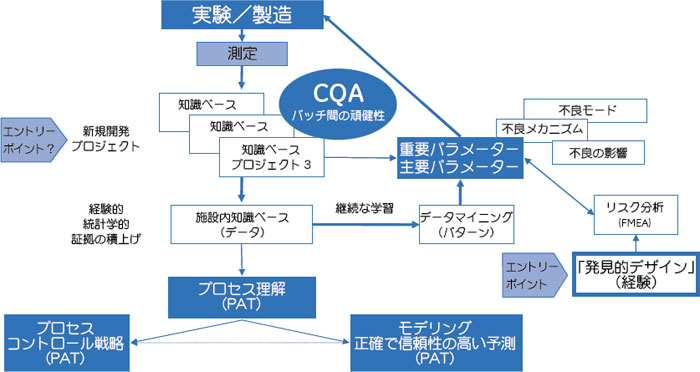
プロセス理解-全体像それにもかかわらず、業界の一部のスポークスマンは「プロセス理解の意識は低く、ましてやPATから得られる重要な利点に関しては理解されていない」(IFPAC 2004、デュポン社、Joe Andrisani氏)と考えています。初期の議論では、しばしば分析ツールキットに別の測定を加えることが言及されていました。しかし、PATは、プロセスの細かな側面の改善をするものでも、不良プロセスのよりよい管理をするものでもありません。PATはより大局的な視点に立つものであり、原材料を効率良くかつ確実に最高品質の製品に加工することを保証するために、製造プロセス全体を包括的な方法でデザイン、モニター、管理するというものです。つまり、PATは製造プロセスのモニタリング、分析および管理によってプロセス性能および製品の品質を至適化するためのシステムなのです。 PATにより効率的なプラットフォームテクノロジーを実現PATプロジェクトはこのような考え方に見合うものであるべきで、全体としての大きな有益性に焦点をあわせなければなりません。会社レベルでこの経済的有益性について理解が得られると、実施もまた容易となるでしょう。PATによって最も大きな財政的利益が得られるプロセスステップを選択することが、着手するステップを決定する際の簡単な経験則です。例えば、あるカテゴリーの対象物質について概念が確立された「プラットフォーム技術」(例えば、モノクローナル抗体またはプラスミドDNAに対する精製プラットフォーム)とPATは明確に結びつけることができます。このような場合、PATはプラットフォームを最新で、効率的で一般的基盤として適用できるよう保つのに理想的なツールです。PATの原則に基づく統合されたモノクローナル抗体プロセスについて言うなら、同じモノクローナル抗体製造のバッチ間だけでなく、他のモノクローナル抗体を精製する新規製造プロセス開発の際にも頑健なプロセスとして取り入れることができるということです。 いずれにせよ、「簡単な仕事」を無視してはいけません。それらは、PATにおいて短期間で利益になり、容易に回収できます。そういった可能性の多くは現行の自動化、センサー、モデリングと関係があり、PATを導入する上で簡潔な方法のひとつです(図5の例を参照)。 PAT戦略の開発と導入PATの中心となるプロセス理解は、製造の初期および後期段階を通じて発展する複雑なプロセスです。 ほとんどの場合、社内外の経験は豊富にあり、(過去の経験に基づく)発見的解決法を通じて、頑健性のあるプロセスデザインへの適当な近道が見つかります。続いて、一連の実験を実施することで、発見的解決法を補完します。知識ベースが確立されるに従い、製品の重要な品質特性に最も強い影響力を有するプロセスパラメーターに関する理解が深くなっていきます(図2)。
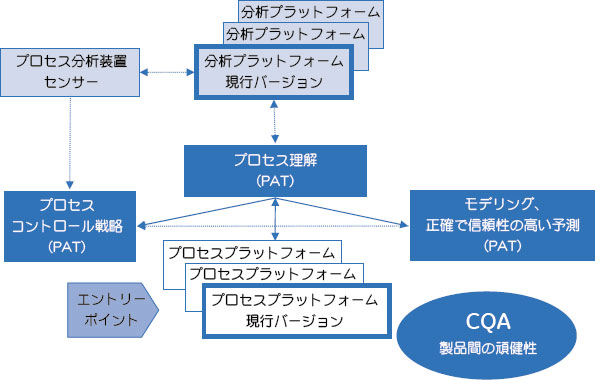
知識ベースにある程度情報が蓄積されると、ついに望んでいた「プロセスを理解している」状態になります。プロセスを理解するためにモデリングを用いた場合、この理解によってその後のプロセス生産量を正確かつ信頼性の高い予測することができ、プロセスコントロールの戦略を明確にすることができます。図3にPAT導入に関する大幅に簡略化したモデルを示します。
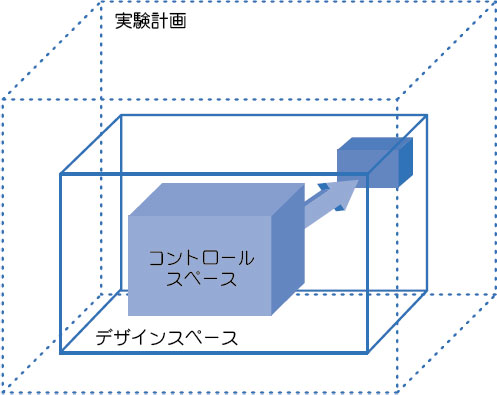
図3. FDAのPATガイダンス文書に示された構想を達成する方法 Quality-by-designとデザインスペース試験によって製品の品質を形成することはできず、製品の品質はデザインによって形成されることを認識するのが重要です。 プロセスを理解するためには、その作業の一部として、プロセスの頑健性の特徴を明らかにし、プロセスバリデーションの計画およびそれを成功させるために必要なデータを集めることになります。こうしてプロセスの特徴を明らかにすると、操作上のパラメーターが変動しても、許容範囲のプロセス性能が得られる「デザインスペース」を設定することができます(図4)。開発およびライフサイクル管理を行っている間の組成や製造プロセスの変更は、デザインスペースを設定するための新しい知識やその他サポート情報が得られる機会となります。
デザインスペースは、製造業者によって計画され、規制評価および承認の対象となります。そのスペース内での操作は変更と見なされません。デザインスペースの境界の外に出た場合は変更と見なされ、通常、承認後変更申請プロセスが開始されます。 リスクベースの品質管理とPATにおけるコントロールPATでは、リアルタイムに継続的な改善を施せるように、プロセスを深く理解し、変動するインプットに対して順応性があることが要求されます。潜在的なリスクを特定する段階では、入力変数を製造プロセスおよびその最終製品のあらゆる面から収集します。これらの因子がプロセスのばらつきや製品の安全性および有効性にどのように影響するかについて、品質管理に対するリスクベースのアプローチで取り組まなければなりません。 リスクマネジメントは現代の品質システムの重要なツールです。たとえば、典型的なProtein Aをベースにしたモノクローナル抗体精製プロセスにおいては、純度に関連するリスクが数多く特定されており、中には回収率が損なわれるものや「規格外」へ直接つながる可能性があるリスクもあります。具体的には、溶出バッファーのpHが低すぎる、溶出用にpHを低くするのが速すぎる、Protein Aカラムに添加した中間体に含まれる不純物プロファイルが不正確であるなどが挙げられます。いずれか、またはこれらすべての例において、Protein Aカラムからの回収画分中の凝集体含量が高くなる可能性があるため、明らかに受け入れがたい結果となります。同様に、回収開始や回収終了のシグナルが適切でない場合は、回収画分中の目的分子の純度が低くなります。 PAT導入時に管理レベルに関して議論する場合、弊社では応答時間によってレベルを以下のように定義しています。
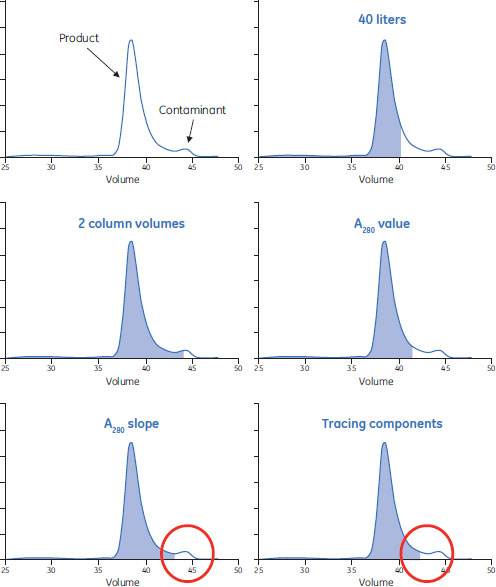
PATのソリューションとしてセンサーテクノロジーを選択する場合、必ずしもOnline/Inlineである必要はありません。FDAのフレームワークのガイダンスでは「タイムリーな測定」とのみ示されていることを覚えておいてください。ただし精製プロセスにおいては、多くの場合、プロセスを今日のペースですすめていくためにも、少なくとも高速なAt-lineの方法の使用が必要とされます。 現行コントロールにおける機会上述のように、現行コントロール法において、短期で容易にPATから利益を得られる点が多く存在します。PAT導入する上で簡単な方法は、自動化、センサーおよびモデリングに着目することです。 例として、ロット間の一貫性が問題となっているとします。この品質特性を保証するためには、あるプロセスから次のプロセスに移行する際(判断ポイント)、明確に規定された基準に基づいて判断されることが必要です。判断ポイントの例として、製品と夾雑物を分離するとき、いつカラムに結合した物質の溶出液の回収を開始するか、または止めるか(分画)というものがあります(図5)。判断基準は多様な形で設定することができます。次のような例が挙げられます。
コントロールパラメーターは、成分検出の記録(すなわち、不必要な物質が溶出され始めた時に示されるUVシグナルと相関するモデル)から選択します。このアプローチは短時間かつ迅速で、容量をベースとした分画よりも望ましく、回収率が落ちるリスクを減らせます。いずれは、直接接続されたIn-lineプローブが、最高のソリューションとなるでしょう。
まとめPATは、重要なプロセスパラメーターと品質特性を測定することによって医薬品製造プロセスをデザイン、分析、管理するメカニズムです。その導入は任意ですが、イニシアティブの一部を導入するだけでも、製造会社にとって取組むに値する経済的利得が得られます。 開発期間の短縮と失敗リスクの低減というプラットフォーム戦略の改善から、最大の利益が得られると思われます(図6)。PATのソリューションはプロセス技術プラットフォームにおいて不可欠な要素となり、特有のソリューションが新たに導入されるたびに、価値が大きくなっていくと確信しています。
下流プロセスにおいて、PATソリューションはバイオ医薬品の損失の回避と、回収率の向上に寄与し、財務上目に見える形で利益を生みます。その結果としてPATの優先順位は高くなることでしょう。中間製品の試験における待機時間を短縮するソリューションにも同様のことが言えるでしょう。直接検出できるプローブの開発および製造での応用によって、プロセス管理戦略の一部として純度のあらゆる側面をより経済的に管理することが可能になります。最後に、リスクベースの品質管理アプローチが台頭してくれば、頑健性を担保する手段としてのバリデーションの重要性は一部失われてゆくでしょう。 参考文献
お問合せフォーム※日本ポールの他事業部取扱い製品(例: 食品・飲料、半導体、化学/石油/ガス )はこちらより各事業部へお問い合わせください。 お問い合わせありがとうございます。 |
||||


© 2026 Cytiva