この記事は、自動翻訳ソフトウェアによって翻訳されています。自動翻訳によって生成された記事(Cytivaにより見直された記事を含む)には、単語、構文、文法などの間違いが含まれている場合があります。弊社は、コンテンツの不正確な翻訳またはその使用により生じる間違いや誤解、または損傷に対して責任を負いかねます。あらかじめご了承ください。
背景
遺伝病やがんなどの治療を目的として、多くの遺伝子治療薬が開発されています。2020年3月現在、世界中で4310件以上の遺伝子治療臨床試験が行われていますが、そのうち第III相および第IV相の臨床試験は518件にとどまっています(1)。遺伝子治療プロジェクトが臨床第III相に入ったり、市場に承認されするにつれ、ウイルスの大量生産が遺伝子治療の産業化の制限要因になっています。
HEK293細胞へのトランスフェクションを用いたアデノ随伴ウイルス(AAV)やその他のウイルスの生産には、静置型システムが依然として最も広く使用されているシステムです。Cell Factory™ システムは、培養ユニットや生産ユニットを追加することでスケールアップが可能ですが、各プレート層と培養液層間のガス交換が一定ではありません。また、細胞の成長を顕微鏡で観察することは非常に困難であり、労働強度、生産コスト、コンタミネーションリスクが大幅に増加します(2)。
浮遊培養は、工業化のための通常かつ簡便な方法です。しかし、細胞の形態、代謝、増殖挙動など、接着細胞と浮遊細胞との本質的な培養プロセスの違いにより、目的産物の品質や収量に大きな影響を与えます。そのため、ウイルス製造のための大規模なHEK293T細胞培養プロセスの確立がますます重要になってきています(3, 4)。
マイクロキャリアは、60~250ミクロンの支持マトリックスで、浮遊系で接着細胞を増殖させることができます。マイクロキャリアは、グルカン、ガラス、その他の合成ポリマーなど、さまざまな材料から作ることができます。これらのマイクロキャリアー材料は、大きな細胞培養面を提供し、高い表面積対体積比を実現します。例えば、5mgのCytodexの表面積は30cm2です。つまり、50Lのバイオリアクターに3g/LのCytodexを使用した場合、単層で培養された10層の細胞工場94個分に相当する表面積が得られることになります。接着培養と浮遊培養の長所を併せ持つマイクロキャリア培養は、細胞を接着させた状態で浮遊培養することが可能です。さらに、大容量の装置を必要とせず、大量の細胞を生産することができます。また、マイクロキャリア培養は、スケーラブルなシングルユースバイオリアクターで行うことができるため、労働負荷やコンタミネーションのリスクを軽減することができます(5)。
Obio Technologyの科学者たちは、接着性のHEK293細胞株を、シェーカーフラスコ内で動物性成分を含まない無血清懸濁液条件下での増殖に成功しました。AAVの生産において、接着培養、懸濁培養、マイクロキャリア培養を比較した結果、マイクロキャリア培養は将来の遺伝子治療製造プロセスをサポートするウイルスベクターの大規模生産に適していると考えています。
材料と方法
マイクロキャリアの調製
Cytodex 1 マイクロキャリアは、製造元の説明書に従い水和・滅菌しました。適量のドライCytodex 1マイクロキャリアを、50 mL/g Cytodex 1でD-PBSに水和させました。水和させたマイクロキャリアを同量のD-PBSを用いて1回洗浄し、121℃で60分間オートクレーブ滅菌しました。上清を除去し、同量のD-PBSでマイクロキャリアを洗浄しました。マイクロキャリアが沈殿した後、上清を除去しています。マイクロキャリア1gに対して、10%FBSを含むDMEMを50mL加え、37℃で4時間インキュベートして使用しました。
セル調製
接着細胞は、HEK293Tの凍結バイアルを37℃の水浴中で急速解凍することにより調製しました。遠心分離後、上清を捨て、ペレットを培地で再懸濁し、T-75フラスコに移しました。2日間培養してコンフルエントになったところで、D-PBS洗浄後、細胞解離用リコンビナント酵素で細胞を剥離しています。その後、T-75フラスコを室温で約2分間静置しました。これらの細胞株では、T字フラスコ拡大プロセスにおいて、1:4から1:6のサブカルチャー比が最も多く適用されました。
懸濁培養と化学的に定義された動物成分無添加培地に同時に適応させたHEK293T細胞から、培地の割合を徐々に増やし、血清濃度を下げる多段階法で懸濁培養細胞を調製しました。得られたHEK293T細胞を無血清培地中でシェイクフラスコを用いて培養しました。
細胞培養
マイクロキャリア培養には、調製したCytodex 1の適量を、125 mLの培地を入れたスピナーフラスコに4 × 105 cells/mLでHEK293T細胞を植え付けました。最終的なCytodex 1の濃度は3g/Lです。
接着培養では、HEK293T細胞を4 × 105 cells/mLでT-25フラスコに5mL培地とともに接種しました。接種後24時間経過し、細胞コンフルエンスが70%を超えた時点でAAVトランスフェクションを実施しました。
浮遊培養では、HEK293T細胞を20mLの培地を入れた振盪フラスコに2×106 cells/mLで接種しました。AAVトランスフェクションは、接種後1〜4時間後に実施しました。
表1に、マイクロキャリア培養、接着培養、懸濁培養の各プロセスのパラメータを示します。
表1.3つの培養プロセスのパラメータ
| 培養の段階 | パラメータ | 培養プロセス | ||
| マイクロキャリア | 接着 | 懸濁 | ||
| 細胞培養 | 培養容器 | Spinner 125 | T-25 | Flask 125 |
| 培養量(mL) | 125 | 5 | 20 | |
| 接種細胞密度 (× 106 cells/mL) | 0.4 | 0.4 | 2 | |
| マイクロキャリア濃度 (g/L) | 3 | NA | NA | |
| 培養時間 (h) | 24 | 24 | 1–4 | |
| トランスフェクション | トランスフェクション培地 | DMEM | DMEM | Opti-MEM™ |
| バックボーン:エンベロープ:ターゲット:PEI (µg/mL) | 1:0.5:1:2 | 1:0.5:1:2 | 1:0.5:1:2 | |
| トランスフェクション後の培地交換時間 (h) | 6–10 | 6–10 | NA | |
| ハーベスト | ハーベストまでの時間 (h) | 96 | 96 | 96 |
| 上清や細胞のハーベスト | Both | Both | Both | |
NAは非該当です。PEIはポリエチレンイミンです。
AAVトランスフェクション
3つのプロセスすべての細胞に、緑色蛍光タンパク質を発現するアデノ随伴ウイルスベクター(AAV2-GFP)をトランスフェクションしました。
マイクロキャリア上の細胞密度が1-1.5×106 cells/mLに達した時点で、スピナーフラスコのスターラーを停止してマイクロキャリアを沈降させました。その後、80%の上澄み液を注意深くデカンテーションし、60%の新鮮な完全培地をマイクロキャリアに添加しました。プラスミドとPEIの混合物を調製するために、10mLの無血清培地を2本の遠心チューブにそれぞれ添加しています。ターゲット、エンベロープ、バックボーンプラスミドを1:0.5:1(μg/mL)の割合で1本のチューブに添加し、もう一方のチューブには、トランスフェクション試薬PEIを適量添加しました。プラスミド混合物を5分間放置した後、PEIチューブの中身をプラスミド:PEIの合計が1:1(μg/mL)になるように添加しました。混合し、8~12分間静置した後、結合したチューブの内容物をスピナーフラスコに加えています。トランスフェクションから6~8時間後に培地交換を行いました。
接着工程では、80%の完全培地を上清と交換し、20%のDMEMを使用してトランスフェクション溶液を調製しました。手順は、マイクロキャリア培養のトランスフェクションと同じです。培地交換はトランスフェクション後6〜8時間後に行いました。
懸濁工程では、10%培養量のOpti-MEM培地をトランスフェクション用培地として使用しました。トランスフェクション後の培地交換を行わない以外は、マイクロキャリア培養のトランスフェクションと同じ手順で行いました。
3つの培養工程におけるトランスフェクションパラメータを表1に示しています。
AAVのハーベスト
マイクロキャリア処理では、トランスフェクションから96時間後に細胞を遠心管にハーベストしました。8~12分間静置し、上清と細胞(マイクロキャリア上)を分離しました。細胞は同量のD-PBSに再懸濁しています。細胞懸濁液(マイクロキャリア付き)は、凍結融解を3サイクル行った後、短時間遠心して細胞の残骸を除去しました。Benzonase™ nuclease (Mg2+)により37℃、2時間消化しました。遠心分離後、細胞および上清の両方について、定量的PCR(qPCR)により力価を測定しています。
接着プロセスでは、トランスフェクションから96時間後に上清をハーベストし、同量のD-PBSに細胞を懸濁させました。そこからは、マイクロキャリアプロセスと同じ手順で行いました。
懸濁プロセスでは、トランスフェクションから96時間後に細胞および培地をハーベストし、同量のD-PBSに細胞を懸濁させました。そこからは、マイクロキャリアプロセスと同じ手順で行いました。
3つの培養工程におけるパラメータを表1に示しています。
バイオリアクタースケールアッププロセス確認
1LのバイオリアクターでAAVを製造するためのパラメータを確認しました(表2)。
表2.バイオリアクター培養のためのパラメータ
| 攪拌回数 (rpm) | 曝気量 | 温度 (℃) | DO (%) | pH |
| 62.5 ± 2.5 | 圧縮空気:2% (12 mL/min), 酸素:自動化 |
37 | 50 | 7.2 ± 0.15 |
結果
マイクロキャリア培養と接着培養におけるHEK293T細胞の増殖速度はほぼ同じであり、倍加時間は約24時間でした。マイクロキャリア培養では、Tフラスコと比較して細胞密度が高くなりました。3つの培養工程におけるトランスフェクション後のHEK293T細胞の形態を表3に示します。
表3.3つの培養過程における細胞形態とAAV2-GFPレベル
| 培養プロセス | 光学顕微鏡 | AAV2-GFP |
| 接着 |  |
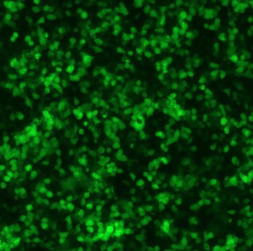 |
| 懸濁 | 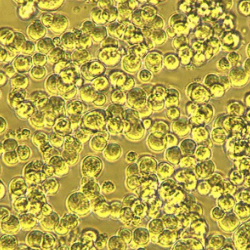 |
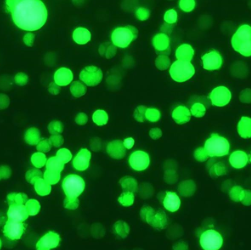 |
| マイクロキャリア (スピナー) |
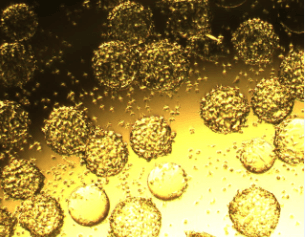 |
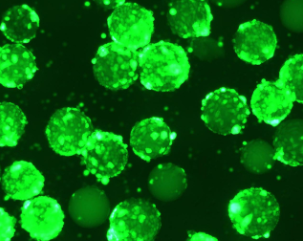 |
3つの培養物すべてについて、細胞および上清のAAV力価を図1および表4で示します。
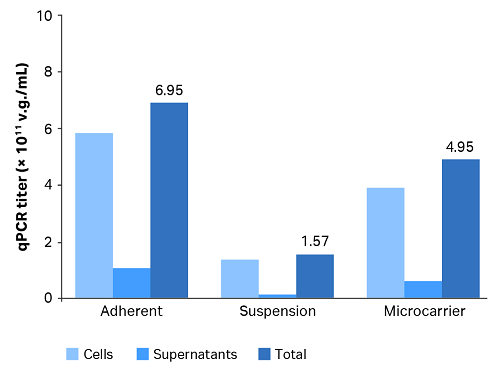
図1. AAVの産生力価。
表4. AAVの産生力価。
| 培養プロセス | qPCR 力価(v.g./mL) | ||
| 細胞 | 上清 | 合計 | |
| 接着 | 5.87 × 1011 | 1.08 × 1011 | 6.95 × 1011 |
| 懸濁 | 1.40 × 1011 | 1.67 × 1010 | 1.57 × 1011 |
| マイクロキャリア | 3.95 × 1011 | 6.44 × 1010 | 4.59 × 1011 |
マイクロキャリア培養と接着培養の合計力価は、浮遊培養の合計力価よりもそれぞれ約2.9倍と4.4倍高くなりました。
マイクロキャリア培養プロセスは、1 L バイオリアクター培養試験で確認されました。主要なパラメータとAAV力価曲線をそれぞれ表5と図2に示します。培養時間が長くなるにつれて、上清と細胞の力価も上昇しました。その傾向は、細胞の曲線においてより顕著であり、96時間では3.3 × 1011 v.g./mLまで、120時間では4.5 × 1011 v.g./mL (qPCRにより決定)でした。
表5.1Lバイオリアクターでのマイクロキャリアプロセス
| 培養の段階 | 条件 | パラメータ |
| 細胞培養 | 接種密度 (× 106 cells/mL) | 0.4 |
| 攪拌回数 (rpm) | 45 | |
| 培養量 (mL) | 400 | |
| マイクロキャリア濃度 (g/L) | 3 | |
| 細胞培養時間 (h) | 24 | |
| トランスフェクション | バックボーン:エンベロープ:ターゲット:PEI (µg/mL) | 1:0.5:1:2 |
| トランスフェクション後の培地交換時間 (h) | 6–8 | |
| ハーベスト | ハーベストまでの時間 (h) | 48, 72, 96, 120 |
| 上清や細胞のハーベスト | Both |
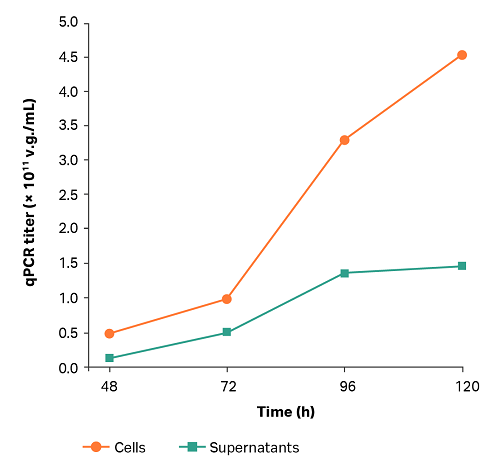
Fig 2.1Lのバイオリアクターでマイクロキャリア処理を行った場合のAAV力価。
結論
これらの結果は、スピナーフラスコと1L攪拌タンク式バイオリアクターの両方で、マイクロキャリア上に高いAAV力価を達成できることを実証しています。さらに、開発したマイクロキャリアプロセスが大規模なAAV生産に適していることを示唆するデータも得られました。
謝辞
Obio Technology社のYongfa Lan氏、HaiRui Yang氏、QingRui You氏の研究成果を紹介する許可をいただき、感謝いたします。
参考文献
- ClinicalTrials.gov database. NIH U.S. National Library of Medicine. https://www.clinicaltrials.gov/ct2/home. Accessed June 16, 2020.
- Yang J, Guertin P, Jia G, Lv Z, Yang H, Ju D. Large-scale microcarrier culture of HEK293T cells and Vero cells in single-use bioreactors. AMB Express. 2019;9(1). doi: 10.1186/s13568-019-0794-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6534633/
- Wang D, Tai PW, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358-378. doi: 10.1038/s41573-019-0012-9. https://www.nature.com/articles/s41573-019-0012-9
- Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31(4):317-334. doi: 10.1007/s40259-017-0234-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5548848/
- Chen XY, Chen JY, Tong XM, Mei JG, Chen YF, Mou XZ. Recent advances in the use of microcarriers for cell cultures and their ex vivo and in vivo application. Biotechnol Lett. 2020;42:1-10. doi: 10.1007/s10529-019-02738-7. https://pubmed.ncbi.nlm.nih.gov/31602549/
お問合せフォーム
※日本ポールの他事業部取扱い製品(例: 食品・飲料、半導体、化学/石油/ガス )はこちらより各事業部へお問い合わせください。