はじめに
2022年、USPは医薬品およびバイオ医薬品の製造に使用されるプラスチック部材の流体接触部から溶出するExtractables(抽出物)に関する要件を規定する<665>章と<1665>章の第4版および最終稿を発行しました。これは、業界の標準化とベストプラクティスの統一に向けた大きなマイルストーンであり、品質の向上、リソースの節約、患者治療に不可欠なマテリアルの適格性確認の迅速化を実現します。Extractablesプロファイルの標準化に向けたこのような進展は、遺伝子療法や細胞療法などの新たな治療法の迅速な開発・展開にとって重要なシングルユースマテリアルのリスク評価を簡素化するものです。
ここでは、標準化されたExtractablesのデータセットに期待すること、および包括的なE & L(Extractables and Leachables、抽出物・浸出物)リスク評価の戦略を支援するためにこれらデータセットをどのように適用するのか、また、追加のシミュレーションやLeachables試験が必要になるのはどのようなケースか、といった点について説明します。 さらに、データセットの標準化の成功が、最終的に患者の安全性リスク評価をサポートするデータの品質と、ラボデータの再現性をさらに向上させる新たな課題への取り組みに対してどのように役立っているかについて、焦点を当てます。
Extractablesの標準化によるLeachablesのリスク評価の簡素化
サプライヤーデータを用いたシングルユースの生物学的安全性評価の出発点は、(i)構造材料および サプライチェーン管理に関する一般的情報、(ii)生物学的反応性データ、および(iii) Extractablesから構成されるサプライヤーデータセットです。Extractablesデータセットは、製造工程で使用する機器から医薬品製造工程へ移行する可能性がある化学物質の特徴を明らかにすることを目的としており、医薬品製造工程への影響(例:細胞の増殖)や製品の品質への影響(例:製剤との反応)、および医薬品最終製剤中に残留して患者の安全性に潜在的な懸念をもたらす可能性があります。
過去10年間で、シングルユースコンポーネントに対するサプライヤーのExtractablesデータの改善への期待が高まるとともに、バイオプロセスの幅広いアプリケーションで使用されるシングルユースシステムの有意義なリスクアセスメントをサポートするためのデータを確実に使用できるよう、業界全体の調整を図るという大きな進展がありました。 2020年には、BioPhorumのシングルユースコンポーネントのExtractablesに関するベストプラクティスの推奨事項の改訂版が発表され、プラスチックコンポーネントのバイオプロセス用部材のUSP <665>の4回目の最終版が発表されました[1]。 USPが2022年に発表した一般通知では、USP <665>の最終的な実施時期を2026年5月と定めており、これは業界に十分な猶予期間を与えることを意図しています。 今日、規制当局は高分子バイオプロセス装置に由来するExtractablesへの配慮を強く認識しており、新規申請時の必要事項の一つとして、シングルユースシステムの評価方法を確認することが一般的となっています。
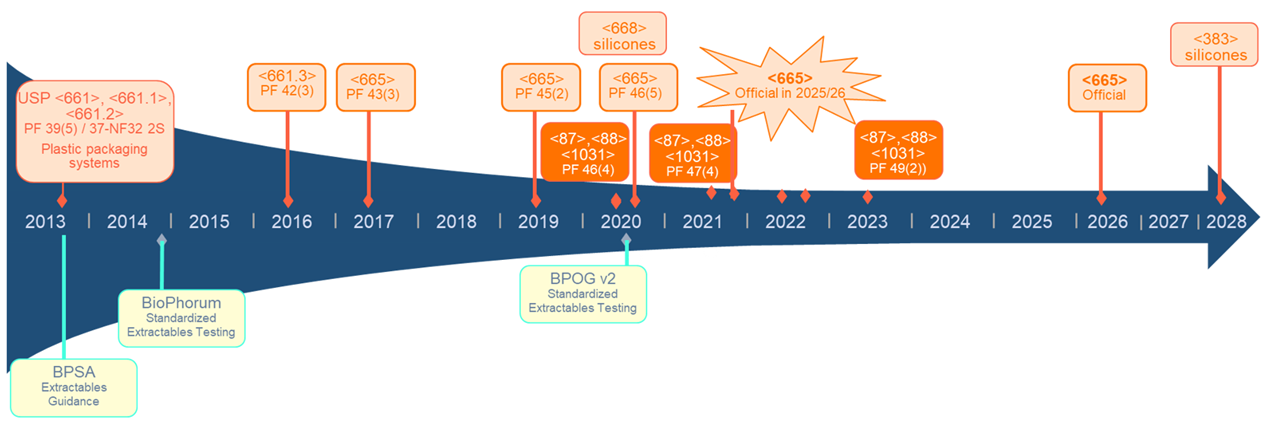
図 1. Extractablesの標準化とシングルユースバイオプロセッシングへの期待に関連するスケジュールの概要
USP <665>
USP <665>と、BioPhorumのExtractablesのベストプラクティスガイダンス改訂版は、コンポーネントの名称、試験間隔の統一、試験方法に対する期待の面でおおよそ一致しており、溶出溶媒のポートフォリオに関してはほぼ一致しています。
USP <665>と、それに付随するUSP <1665>ガイダンスの章は、USP <665>への準拠が単にデータが入手可能であることを示すものの、事前に確立された安全性の許容基準を満たすことを示すものではないという点で、歴史的なpass/failの章とは異なります。 この試験の目的は、バイオプロセスで使用するシングルユースシステムが、特性を十分に評価したコンポーネントと材料から設計され、なおかつ設計による品質と品質リスク管理アプローチを強化することにあります。シングルユースシステムを医薬品製造工程へ使用するための適格性の判断は、製薬会社の評価担当者の責任となります。
さらにUSP <665>では、シングルユースシステムを使用するバイオプロセッシングアプリケーションには、低リスク、中程度のリスク、高リスクのものがあり、リスクが高いアプリケーションほど、より包括的なデータによる支援が一般に必要になるとの認識が示されています。 USP <1665>は、低リスク、中リスク、高リスクの判断のためのガイダンスを提供しています。 低レベルのデータセットでは、50%エタノール/水による溶出に続き、不揮発性蒸発残渣物(NVR)やUV吸光度のような化合物の非特異的な測定が行われます。中程度のリスクレベルのプロファイルでは、50%エタノール/水での溶出のみが行われますが、通常、ヘッドスペースGC/MS、直接注入GC/MS、LC/UV/MSによる有機化合物の化合物固有の分析が必要とされます。 高リスクレベルのプロファイルには、中程度の溶出プロファイルに加え、特定の低pH(pH3)および高pH(pH10)Extractablesにおける化合物特異的プロファイル、ならびに通常ICP/MSによって評価される元素不純物の評価が必要となります[2]。 またUSPでは、低pHにおけるリスクについて、BioPhorumの0.1M H3PO4低pH溶出溶媒を代替溶媒として評価を行えることに言及しています。
BioPhorumのExtractablesのベストプラクティス
BioPhorumのアプローチはUSPと類似していますが、pH範囲がより広く 〔0.1M H3PO4 (~pH 1.6)、および0.5N NaOH (~pH 13.5)〕、試験間隔の追加が求められ、さらに水による溶出プロファイルの要件が追加されています。
BioPhorumのアプローチでは、USP <665>の中程度リスクレベルのプロファイルと同様に、50%エタノール/水を単一溶媒として低リスクの低分子成分を溶出することも可能です。
水による溶出プロファイルは、USP <665>の高リスクレベルプロファイルのサブセットであると予想されますが、多くのバイオプロセスアプリケーションとの類似性が付加価値の提供となる可能性があり、そのような場合は、エンドユーザーのリスク評価を大幅に簡素化することができます。 USPは、BioPhorumの高pH溶媒を理由なく代替条件とすることを認めていません。それは、より高いpHのBioPhorum 0.5N NaOHの場合、ほとんどの適用条件で典型的とは言い難い、より複雑なプロファイルを作り出す可能性があるためです。USPはさらに、pHが非常に高いとき(< pH 10)に通常予測される溶出特性が、大半のアプリケーションに完全に適している革新的な材料にとって、不利なものとなる可能性があることを示唆しています。 したがって、BioPhorumの高pH溶媒を使用する場合は、USPに準拠するための根拠が必要となります。
サプライヤーデータへの期待
現在、化合物固有の溶出プロファイルが業界内で強く連携していることから、エンドユーザーはUSP <665>の低リスクプロファイル、中リスクプロファイル、高リスクプロファイル、またはBioPhorumに沿ったプロファイルの取得を期待することができます。 ほとんどのサプライヤーは、高リスクの用途に使用される主要成分のデータを既に保持しており、その他の成分についてもUSP <665>で要求されるデータの作成プログラムを導入しています。このプログラムは、市場に投入された新規のコンポーネントや、重要なアプリケーションに関係するコンポーネントが重視されます。
USP <665>データ適用の可能性を検討する場合、
- データのリスクが低レベルなのか、中レベルなのか、高レベルなのかを明確にすべきである。
- 溶出前の電離放射線処理やフラッシングなどの前処理条件についても、溶出プロファイルとして最終使用条件を示す必要があるため、注意するべきである。オートクレーブ滅菌後のシングルユースコンポーネントの使用も可能であるため、加熱滅菌後のデータセットも数多く存在する。しかし、照射後と加熱前処理後では、一般にプロファイルは異なる。
- 同一の構造材料で作成され、同一の工程で製造された(すなわち、同じコンポーネントファミリーに属する)類似コンポーネントの部品番号のリストを提供する必要がある。このリストは、より小さな、またはより大きなコンポーネントサイズを識別するスケーリングデータに適用するパラメータ(表面積など)を示す。
- データのライフサイクル管理は、データ報告以降にサプライヤーのマテリアルまたは製造プロセスに変更があった場合の評価を含めてレビューする必要がある。
シングルユースアッセンブリについては、アセンブリに使用するマテリアルリスト、関連するExtractables報告書、およびデータのスケーリングに関連する部材のデータも提供する必要があります。
その他の規格
今日の業界は成熟とともに<665>データへの期待に起因する混乱があり、他の規格も考慮する必要があるかもしれません。
- 医薬品最終包装材として使用するプラスチック容器には、従来USP <661>が適用されていましたが、USP <665>の発行と並行して大幅な改訂が行われました。従来のUSP <661>では水による溶出前にプラスチック容器を3回すすぐことを要求していましたが、現在この章は実質的に廃止されています。 その代わりとなる章には、医薬品の最終包装材に使用するプラスチック(容器施栓系)を評価する標準的解析に焦点を当てた<661.1>、医薬品の最終包装システムに固有の <661.2>があります。 しかし、USP <665>が発行されたことから、これらの章はシングルユースアセンブリには適用されないと予想されます。
- USP <383>はシリコン材料を対象とした新しい章で[3]、<661>および<665>への改訂に伴って発行され、EP 3.1.9のシリコンに関する章と密接に関連しています。シリコンはバイオプロセスコンポーネント(例:チューブ、ガスケット)、またはその一部として使用できることから、USP <665>でプロファイリングされることが期待され、ガスケットやシールなど<665>の評価対象ではない他のシリコン材料は、<383>に従って評価される可能性があります。 なお、シリコンのプロファイリングを複数の規格に従って評価することは想定されていません。
標準データに対する規制上の期待と要件
USP <665>は現在、2026年5月に正式に制定される予定です[4]。USP-NFで認可・商品化された医薬品が、米国食品医薬品化粧品法(FD&C法)公認のUSPに準拠するには、医薬品モノグラフ、一般章および通知で示されている要件を満たす必要があります。しかし、USP <665>は現在、特定のUSP医薬品のモノグラフ、一般章および通知の必須要件ではないため、USP準拠の目的で適用可能な必須要件は、今のところ存在しません[4][5]。USP <665>の正式発行延期の目的は、<665>を適用要件とすることをさらに深く検討し、ICH Q3E開発グループの動向と並行して追跡することで、業界が適用する時間的余裕を確保することです。
FDAはUSP <665>の開発プロセスに積極的に参加し、バイオ医薬品の非モノグラフ規格を頻繁にサポートしています[6]。したがって、USP <665>はFDAの公式認定は受けていないものの、流体が接触する工程の機器に対するベースライン評価の方向性が固まりつつあり、それに対する期待が高まっていると考えられています。2024年初めに実施されたバイオ医薬品業界のグループに属する主要な専門家を対象とした調査では、回答者の少なくとも85%がUSP <665>が全ての新規申請に対する要求事項であると認識していることが強調されており、FDAや他の規制当局がシングルユースシステムをどのようにリスク評価するかを、申請に対して示すことを求めているとの回答もあります。業界内で最も理解を得やすく協調し得る対応は、USP <665>に従うことへの言及であり、これは広く受け入れられているようです。もちろん、非典型的あるいは重要なアプリケーションで使用されるシングルユースコンポーネントには、さらなる評価が必要となるでしょう。
ExtractablesとLeachablesのリスク評価
シングルユースアセンブリのリスクアセスメントは、一般的に、プロセス開発初期の機器選定段階、および全てのプロセス条件をより具体的に設定する正式な適格性確認段階で実施されます。いずれの場合も、全体的なアプローチは同じです。
選定フェーズ
選択フェーズにおいて、エンドユーザーは、コンポーネントまたはシステムのリスクレベルを評価し、Extractablesに関するサポートデータ(通常サプライヤーから提供される)がリスクレベルに適合していることを確認します。サポートデータからは、一般的な品質を評価し、医薬品または製造工程に影響を与えることが知られている被験物を特定して、安全性リスク評価をサポートするための合理的な情報、および特定された高リスクレベルの化合を確認することができます。しかしながら、マテリアル選定の時点では、最終的な工程パラメータの設定、あるいは最終的な分析評価閾値設定のための工程が十分に開発されていない場合があると一般に理解されています。
シングルユースシステムに対する業界内で統一されたリスク評価アプローチ
多くの複雑ないくつかのの部材から構成されるアッセンブリの評価を簡素化することを目的として、業界内で統一されたExtractables & Leachables(E&L)リスク評価ツールが作成されています。最も広く使用されているモデルとしてBioPhorum[7]やELSIE[8]が挙がり、今日ではUSP <1665>[9]が加わりました。 BioPhorumのアプローチはシングルユースアセンブリのアプリケーションに特化したもので、リスクランキングの計算に基づいて、3つのリスクレベル(低、中、高)のいずれかを出力します。
ELSIEモデルは、包装と同様に製造およびドラッグデリバリーシステムを包含し、FEMA(確率とリスクレベルを表す4x4のマトリックス)に基づいて、使用者の選択によって2レベル(低または高)または3レベル(低、中、高)のいずれかのスコアを出力します。USP <1665>のアプローチは、プラスチックのバイオプロセス構成部材に焦点を当て、4つの主要なリスク領域と個別の緩和要因を考慮して、3つのリスクレベル(低、中、高)のいずれかを出力します。
シングルユースシステムの適格性評価を目的としたE&L評価の実施
各企業のリスクアセスメントモデルと適格性評価プロセスにより、必要とする具体的な情報は若干異なる可能性があります。全体としてみると、医薬品製造工程におけるシングルユースシステムのリスク評価と、適格性評価に必要となる共通の前提条件の情報があります。これには、(i)医薬品製造工程で使用するすべてのシングルユースシステムについて完璧に理解するためのエンドツーエンドの工程フロー図、(ii)工程条件(例えば、温度、接液時間、流体マトリックス、工程の容量など)、(iii) シングルユースシステムおよび構成部材の情報(構成部材のリスト、ポリマーの種類、部品識別番号、製造元、およびポリマー樹脂コード(入手可能な場合)、(iv)滅菌または前処理方法、および(v)投与経路および投与方法、が含まれます[10]。
標準データを用いた部材の評価
USP <665>やBiophorumに準拠し、標準化した成分のExtractablesデータセットが利用可能になりつつありますが、これらのデータセットをどのようにアセンブリ評価に適用するかの具体的な手順については、明確ではないかもしれません。 最も直接的なアプローチは、報告限界値と半定量法を考慮しつつ、アセンブリを構成する各コンポーネントのサイズと数に合わせて試験済みのコンポーネントデータをスケーリングする手法です。関連溶媒の化合物をスケーリングして加算するこのアプローチは、単純ではありますが、特に多数の化合物を報告する場合や、異なる研究室で生成されたデータセットに依存する場合、多くの時間を要する複雑な作業になる可能性があります。
重要なコンポーネントの評価のための代替的なアプローチとして、各コンポーネント評価のための閾値(AETcomp)を、アセンブリの閾値(AETassembly, mcg/ml)とプロセスの容量(Vprocess)の積として計算し、コンポーネントの表面積(SAcomp) と不確定係数(UF)で除する方法があります。 同じ化合物が異なるコンポーネントから検出される場合には注意が必要ですが、多くの、あるいはすべてのコンポーネントがAETcompよりはるかに低い値となる可能性があることから、このアプローチはデータセットの評価を簡素化すると考えられます。同手法はまた、コンポーネントデータセットの報告限界値の妥当性の確認においても有用です。
適格性評価フェーズ
適格性評価の際、リスク評価者は既存の溶出プロファイルが製造工程の条件を適切に反映しているかどうかを検証し、既存のExtractablesデータの定量方法に関連する安全調整係数を考慮して、AETを超える化合物に評価を集中させます。評価者は、状況に応じてデータのレベルがプロセスを適切に反映し、なお且つ安全性リスクアセスメントを結論づけるための有効性について、合理的な判断を行います。 既存データがプロセス条件を十分に反映していない場合、または標準データを積極的に収集して評価対象が膨大になった場合は、工程ごとにPERL(process equipment related leachables)シミュレーションを行い、分析上実用的な溶媒/溶液を使用して、特定の医薬品製造プロセス条件の正確な適応を推奨します。 状況に応じて、実プロセス流体および工程条件によるLeachables評価の実施も視野に、これらの対応によるリスク評価の対象化合物の数をさらに減らすことができると、一般に期待されています。
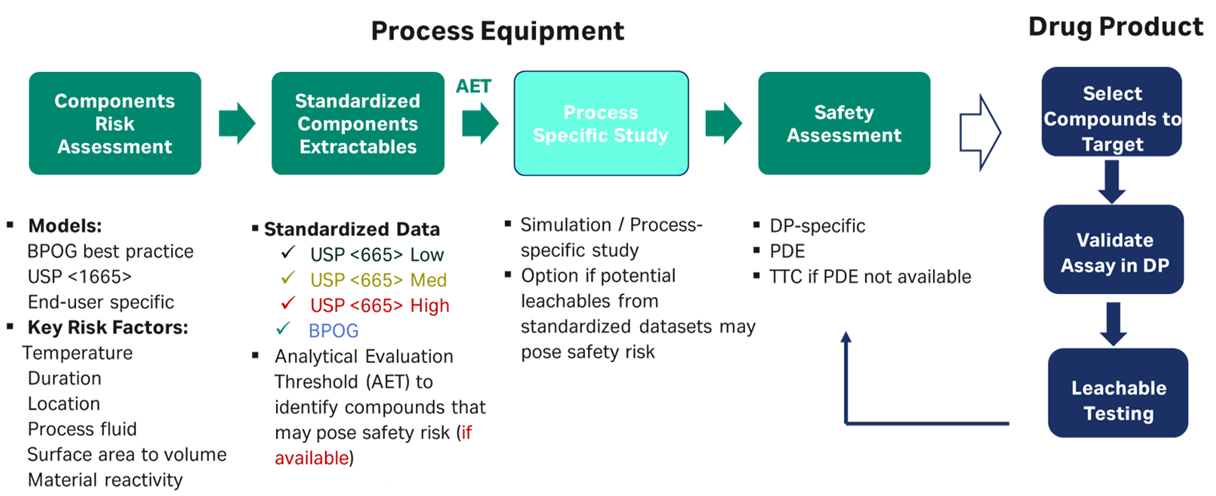
図 2. シングルユースアセンブリの包括的なE&Lリスクアセスメント。 医薬品(DP)、一日許容曝露量(PDE)、毒性学的懸念の閾値(TTC)。
新たなトレンドと進化するトレンド
シングルユースコンポーネントの溶出プロトコルの標準化が大きく進展したことで、現在、(i)すべてのバイオプロセスコンポーネントデータの取得に要する時間、(ii)データを取得するラボ間でデータの一貫性と品質を向上させる取り組み、(iii)標準的なExtractablesデータを用いたリスク評価は、ある種のケースでは適用可能であるということへの理解、(iv)標準データは新たな課題への対処にどのように役立つのか、(v)将来のICH Q3Eガイダンスが今日の実務にどのような影響を与えるのか、などへ関心が移っています。
シングルユースの将来
これまでもExtractablesデータの標準化に対して注目されていましたが、それは主に、シングルユースアセンブリに組み込まれたコンポーネントに焦点を当てたものでした。2026年5月に公式発行予定のUSP <665>では、カートリッジフィルターや加熱滅菌アプリケーションでは使用されるものの、アセンブリに含まれていなかったバイオ製造コンポーネントもカバーするよう、範囲の拡大が予想されます。この範囲の拡大は、すべてのデータが利用可能になるまで時間がかかるものの、最終的にサプライヤーのデータに対してどのような期待が持てるのか、という共通の目標を明確に設定するものです。
ラボデータの一貫性と品質の改善
Extractable試験の標準化が進むにつれ、異なるラボにおいて、同一のコンポーネントを同一条件下で試験したデータセットを目にすることが多くなりました。 従来、コストおよび特定の試験設計の面から、これは非常に珍しい状況でした。この状況は、検出されるべき化合物の報告漏れ(省略の誤り)や、評価対象サンプルや化合物に関する経験が浅いラボでは、化合物の同定や定量が不十分である可能性を示唆しています[11]。高解像度質量分析能を有するLC/MSを自社で利用できることへの期待が高まり、また化合物の同定と、同定された化合物の特定の解決に役立つGC/MSの必要性が高まるにつれて品質に対する期待も高まり、ラボがその分析能力の範囲内で化合物の強力なワーキングライブラリを保有することが求められています。
2023年、USPはラボでExtractable & Leachables評価に使用するクロマトグラフィー法および装置の適合性評価に使用する外部基準化合物のリストを示す刺激的な論文[12]を発行しました。これには、ヘッドスペースGC/MS、直接注入GC/MS、エレクトロスプレーLC/MS、およびAPCI LC/MS用の特定の標準混合物が含まれます。これらの化合物の標準品は、一般的なExtractable & Leachables分析方法の分離能と検出能を評価し、業界全体でラボ間の一貫性を高めることを目的としています。 この論文の公開時点で、当初提案された標準試料は、従うべき標準混合物に関する最新の推奨事項とともに、さまざまなラボや機器での評価フェーズを完了しています。
細胞・遺伝子治療に関する考察
細胞および遺伝子治療の成功率が増加するにつれ、シングルユースアッセンブリのバイオ医薬品に関連するリスク評価戦略、および標準化されたExtractableデータセットが、細胞および遺伝子治療の評価に適しているのかという点について、しばしば疑問が呈されています。 ほとんどの場合、USP <665>で規定されているデータセットは、細胞治療や遺伝子治療のアプリケーションを十分に支援し得ると考えられています。一例として、細胞凍結アプリケーションでは、細胞膜を破壊しうる大きな氷晶の生成を防ぐために、しばしばDMSO(ジメチルスルホキシド)が添加されます。誘電率などを使用した溶媒溶出能力の評価に基づくと、BioPhorum および USP で規定されている 50% エタノール/水混合物がこれらの用途に十分対応していること考えられ、得られた実験データもこの結論を裏付けています。これまでのところ、細胞治療アプリケーションの多くは、通常、医療機器市場を支援するためのプロセス固有のISO 11137-18データセットを使用していました。この業界が成熟するにつれて、これらのアプリケーション支援のために使用するマテリアルや溶出プロファイルに関する標準化が進むと期待されています。
細胞療法に関するさらなる課題は、初期の臨床試験用に選択されるマテリアルから生じます。これらのマテリアルは、多くの場合、研究室や病院の保管庫から採取され、必ずしも一般的なバイオ医薬品製造工程のシングルユースアッセンブリ由来ではありません。これらのマテリアルに関する課題は、現在、バイオ医薬品処理機器向けに期待されるデータセットによって十分にサポートできない可能性があり、そのようなマテリアル(PVC など)に対する E&L リスクが高くなる可能性があるという点です。
抗体薬物複合体(ADC)に関する考察
抗体薬物複合体は、潜在的な薬物療法として急成長の分野であり、多くの場合、細胞毒性またはその他のペイロードと抗体との結合を促進するために、DMSO(ジメチルスルホキシド)やDMAC(ジメチルアセトアミド)などの高度な有機溶媒を使用します。これらの有機溶媒は一般的に5~20%程度であり、USP <665>で一般的な50%エタノール/水混合溶媒に匹敵する溶媒溶出力を持ちます。一般にADCを扱う場合はマテリアルの適合性評価とオペレーターの安全確保が最重要課題であり、結合工程に続いて製造工程のクリアランス工程(TFF透析濾過など)が追加されます。これは、医薬品最終製剤に関連する不純物の残留リスクを低減するためです。
ニトロソアミン
2018年以降、1200万本以上の血圧降下剤がニトロソアミンの混入によって市場から排除され、ニトロソアミン混入の可能性を評価する規制要件が推進されました。ニトロソアミンやその他の高リスクが懸念される化合物は、典型的なE&Lスクリーニング法の報告義務の限界値をはるかに下回るレベルでリスクの懸念を引き起こす可能性があるため、一般にExtractableは最初のリスク評価には考慮されません。マテリアルと製造プロセスの条件がニトロソアミンの生成につながり易いか否かが焦点となります。 従って、このリスク評価は一般に、二級または三級アミン源(NMP、DMF、四級アンモニウム塩など)がニトロソ化剤(亜硝酸塩、加硫剤など)と接触するリスクを評価することに依存します。ほとんどの場合、これらのリスクはシングルユースアッセンブリを構成するマテリアルと製造プロセスの設計に基づいて対処することができます。
クリアランス
USP <1665>に記載のリスクアセスメントモデルに基づき、アプリケーションの下流側で発生する有意なクリアランスまたは容量希釈ステップは、全体的なコンポーネントのリスクスコアを低減する役割を果たします。ほとんどの低分子は、透析ろ過によって容易に希釈される傾向があることから、これは論理的な手法と言えます。しかしながら、この結論を支持するために期待されるデータのレベルやクリアランスが、分子の大きさにのみ基づいているのか、または溶解性に関連する他の特性によっても影響を受ける可能性があるのかについては、懸念が残ります。この分野における刊行物の増加は、クリアランスの理論的根拠をさらに裏付けるのに役立っています[13]。
微粒子
医薬品に含まれる微粒子不純物は、患者の安全性に関わる重要なリスクであり、医薬品のリコールの最大要因となります。微粒子はシングルユースや包装材料に関連する可能性があるため、しばしば微粒子を広義的に不溶性のExtractablesと見なすべきか否かが議論されます。さらに、微粒子の典型的な評価方法はFTIR/ATRを使用することであり、従来よりマテリアルとExtractablesを特徴付ける一般的なスクリーニング方法です。しかし一般に、微粒子は製造環境と清浄度に由来する不純物であるのに対し、Extractablesはマテリアルの表面、または内部から医薬品製造プロセス液へ移行する化学的不純物と関連性が高い、という大きな違いがあります。
評価の簡素化
シングルユースアッセンブリからのExtractablesデータセットの標準化が進むにつれ、今日では業界で統一されたデータセットが一般的となり、医薬品プロセスで使用される機器のリスクを評価する能力がこれまで以上に強化されました。 そのため、これらのデータセットがADCや細胞および遺伝子治療といった新しいアプリケーションやモダリティをほぼ網羅しているかどうかという新たな質問にも答えられるようになりました。Extractablesデータ適用の可能性と透明性への期待が高まり続ける中、バイオプロセスE&Lに参入して試験を実施するラボが増加しており、今後も継続的な取り組みによって、さまざまなコンポーネントやラボのデータを意味のある形でまとめ、一貫したプロセスと製品のリスク評価をサポートできるように、報告書の品質と一貫性を確保することを目指します。
まとめ
USPは2022年に医薬品製造に使用されるプラスチックのExtractablesに関する<665>章および<1665>章を発行し、品質向上とリスク評価の簡素化を目指しました。これにより、シングルユースコンポーネントのリスク評価がされ、新たな治療法の迅速な開発が期待されます。さらに、データの一貫性と品質を改善する取り組みが進行中であり、規制当局もこれを強く認識しています。新たなトレンドとして、細胞・遺伝子治療や抗体薬物複合体(ADC)などの新しいアプリケーションに対する評価が進んでいます。
著者が所属するCytivaでは40年以上にわたるバリデーションサポートの実績をもとに、重要工程のフィルターバリデーションやシングルユースシステムの使用に関する多くの現行基準の確立に貢献してきました。また、専門的なコンサルティングおよび試験サービスを提供することで、お客様の速やかな開発をサポートし、患者の安全を確保します。お困りのことがありましたらお気軽にご連絡ください。
Reference
- [1]United States Pharmacopeia, “<665> Plastic components and systems used to manufacture pharmaceutical drug products and biopharmaceutical drug substances and products,” USP, 2021.
- [2]United States Pharmacopoeia, “<232> Elemental Impurities - Limits,” USP, 2020.
- [3]United States Pharmacopoeia, “<383> Cured Silocone Elastomers for Pharmaceutical Packaging and Manufacturing Components,” Pharmacopoeial Forum, vol. 48, no. 3, 2022.
- [4]United States Pharmacopeia, “Notice of Intent to Revise,” 25 Feb 2022. [Online]. Available: https://www.uspnf.com/notices-665-nitr-20220225
- [5]United States Pharmacopeia (USP), “General Notices and Requirements,” USPNF, no. 3, 2022.
- [6]P. Nithyanandan, “FDA-USP Collaboration and Partnership,” 2023.
- [7]BioPhorum, “Best Practices Guide for Evaluating Leachables Risk from Polymeric Single-Use Systems Used in BioPharmaceutical Manufacturing,” BioPhorum, 2018.
- [8]ELSIE, “Risk Assessment for Leachables in Drug Product and Drug Product Manufacturing Systems”.
- [9]United States Pharmacopeia, “<1665> Characterization and qualification of plastic components and systems to manufacture pharmaceutical drug products and biopharmaceutical drug substances and products,” USP, 2021.
- [10]United States Pharmacopeia, “<1663> Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems,” USP.
- [11]P. Christiaens, J.-M. Beusen, P. Verlinde, J. Baeten and D. Jenke, “Identifying and Mitigating Errors in Screening for Organic Extractables and Leachables: Part 1-Introduction to Errors in Chromatographic Screening for Organic Extractables and Leachables and Discussion of the Errors of Omission,” PDA J Pharm Sci Technol, vol. 74, no. 1, pp. 90-107, 2020.
- [12]D. Jenke, J.-M. Beusen, P. Verlinde, J. Baeten, P. Christiaens, D. Hunt and P. Gruddanti, “Proposals for the development, composition, and routine use of system suitability mixtures in support of chromatographic screening for organic extractables and leachables,” USP Pharmacopeia Forum, vol. 49, no. 4, june 2023.
- [13]B. Sun, M. Hadidi, J. Nunez, B. Song, G. Tumambac, K. Wong, G. Kalinowski and J. Hathcock, “Efficiency of ultrafiltration/diafiltration in removing organic and elemental process equipment related leachables from biological therapeutics,” vol. 40, no. 1, 2024.