本文中に出てくるマークの意味
:ご注意 / :プロトコールを変更できる箇所 / :詳しい情報
電気泳動とは
ウェスタンブロッティングのはじめのステップである電気泳動をご紹介します。大小さまざまなタンパク質が含まれるサンプルをサイズの違いに並べることで目的のタンパク質を見つけやすくなります。
電気泳動の原理
タンパク質の電気泳動法としては下記3 つの手法が主流となっています。
- SDS-PAGE:分子量の違いにより分離。
- 等電点電気泳動:タンパク質の等電点(チャージ)の違いにより分離。
- 二次元電気泳動:等電点・分子量の2 つのパラメーターの違いにより分離。一次元目に等 電点電気泳動を、二次元目にはSDS-PAGE を行う。
この中でウェスタンブロッティングでよく用いられているのはSDS-PAGEです。最近は二次元電気泳動で分離したゲルをもとにしたウェスタンブロッティングも多くなってきています。このハンドブックではSDS-PAGE を中心にご紹介していきます。
SDS-PAGE
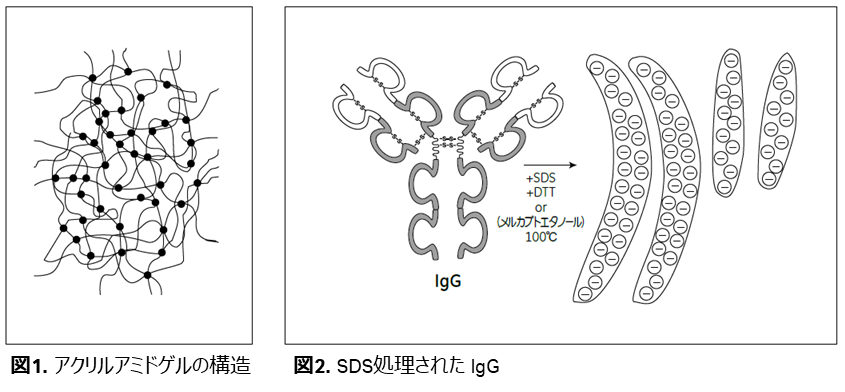
SDS(Sodium Dodecyl Sulphate)- ポリアクリルアミドゲル電気泳動(SDS-PAGE)は、目的タンパク質の高次構造を変性して分子量の違いにより分離する手法です。ポリアクリルアミドゲルは、ゲル中の細孔径が密なため100 ~ 200 kDa 以下のタンパク質やポリペプチドを分離するのに適しています(図1)。操作が簡便で再現性が高いので、タンパク質の電気泳動では最もよく用いられている手法です。通常は、泳動サンプルの調製時にβ - メルカプトエタノールやDTT(Dithiothreitol)などの還元剤を添加してタンパク質のS-S 結合(ジスルフフィド結合)を切断します。SDS は、水溶性タンパク質1 g あたり約1.4 g 結合してSDS-タンパク質複合体を形成します。SDS の結合量によって分子の電荷がほぼ決まるため、電気泳動によりポリペプチド分子を分子量にしたがって分離することができます。(図2) にIgG をSDSとDTT(還元剤)を含むサンプルバッファーに溶解し、100 ℃で処理した例を示します。還元剤によりジスルフィド結合が切断され、IgG はH 鎖とL 鎖に分かれます。さらにSDS が吸着し全体が負に帯電することで、立体構造がなくなり、タンパク質は一本鎖となります。SDS 処理により、タンパク質固有の立体構造や表面電荷違いによる影響を受けることなく、分子量での分離が可能になります。電気泳動により、分子量の小さなタンパク質は速く、大きなタンパク質は遅れて陽極方向に移動します。
SDS-PAGE における目的タンパク質の分離能はタンパク質の大きさとゲルの多孔度に依存します。そのため、最適なゲル条件を見つけるために条件検討が必要となります。
SDS-PAGEに必要なもの
- 電気泳動装置
- ポリアクリルアミドゲル
- サンプルバッファーで処理したサンプル
- 分子量マーカー
- 染色試薬
※ウェスタンブロッティングでは確認用のオプション操作です。
ウェスタンブロッティングでの注意点
- サンプル添加量の調整
希釈系列や添加量を変えてサンプルを泳動します。いずれかの条件を最適条件内に入れることが目的です。 -
還元処理による目的タンパク質の変性
一次抗体が目的タンパク質の立体構造を認識する場合は、還元処理により立体構造が変わり、抗体が反応しなくなる場合があります。このようなときは非還元処理*を行います。
*:泳動前のサンプルの処理時の条件をタンパク質の立体構造を変えない条件に変更すること。DTT、メルカプトエタノールを含まないサンプルバッファーを使用します。また、熱処理も37℃、30 分など平穏な条件にします。温度時間などはサンプルに依存します。 -
有色マーカーの同時泳動
有色分子量マーカーをサンプルと一緒に泳動すると、泳動後のゲルやブロッティング後のメンブレン上にマーカーのバンドが目視で確認できるので、泳動状態やメンブレンへのブロッティング効率のチェックに有効です。(有色マーカーはマーカータンパク質に色素が結合しているので、正確な分子量測定には向きません。分子量の目安として用いてください。) -
ポジティブコントロールやネガティブコントロールの同時泳動
複数のバンドが検出された場合やバンドが全く検出されない場合のトラブルシュートが容易になります。 - 適切なゲル濃度
目的タンパク質が分離ゲルの中央にくるゲル濃度を選択します。
ポリアクリルアミドゲル濃度の選択
分離できる分子量分画範囲はポリアクリルアミドゲルの濃度によって決まります(表1)。精度の高い解析を行うには、目的タンパク質のバンドがゲル中央付近に位置するようにゲル濃度を選択します。ゲルには濃度勾配を持つグラジエントゲルとゲル濃度が均一なホモジニアスゲルがあります。サンプルのおおよその分子量分布を調べる場合、もしくは、サンプルのタンパク質の分子量が広範囲にわたる場合は、グラジエントゲルを用います。サンプルの分子量をできるだけ正確に調べたい場合、あるいは分子量の近いタンパク質を分離したい場合には、濃度が均一なホモジニアスゲルを用います(図3) 。グラジエントゲルとホモジニアスゲルを組み合せることでシステマチックにサンプルの分子量分布を解析できます。
表1.タンパク質分画に必要なポリアクリルアミドゲル濃度
| 分子量範囲(kDa) | 分離ゲルの濃度(%) |
|---|---|
| 36~205 | 5 % |
| 24~205 | 7.5 % |
| 14~205 | 10 % |
| 14~66* | 12.5 % |
| 14~45* | 15 % |
*分子量の大きなタンパク質はゲルに入りにくい場合があります。
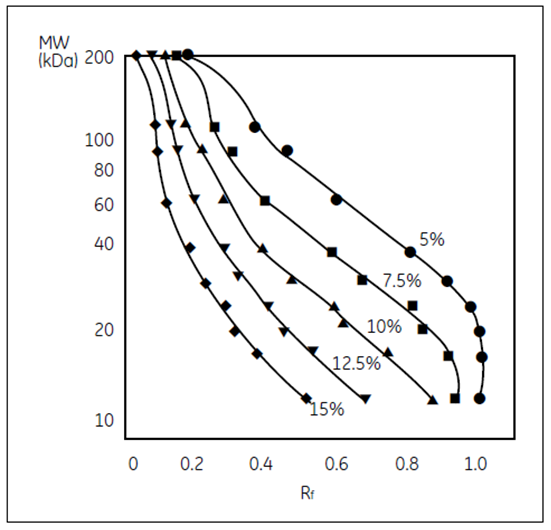
図3.標準タンパク質の移動度とポリアクリルアミド濃度の関係
タンパク質マーカーを5 種類の濃度のポリアクリルアミドゲルでSDS-PAGE を行い、結果をプロットしました。
ポリアクリルアミドゲルの組成はアクリルアミドの割合(% T)とクロスリンクの割合(% C)の2 つの指標で示されます。クロスリンクの割合は通常2.6%です。% T が高くなるとポアサイズが小さくなるのでタンパク質がゲル内で移動しにくくなります。
もっとも一般的なウェスタンブロッティングでは2 種類のゲルを用います。濃縮ゲルと分離ゲルです。濃縮ゲルはホモジニアスなゲルで大きめのポアサイズ(% T=4 ~ 5%)で作製します。これは分離ゲルに入る前にタンパク質を濃縮する役割を果たし、ゲルの中でのバンドの分離能を高めます。一方で分離ゲルはより固いゲルにします。ホモジニアスやグラジエントゲルどちらでも% T は5 ~ 20%の幅で設定します。
分子量の測定法
サンプルと同一ゲルに分子量マーカーを泳動することで、サンプル中のタンパク質の分子量を推定することができます。正確な分子量測定には色素が結合していないマーカーを用います。(図4)は分子量既知のタンパク質分子量マーカーをSDS-PAGE した結果です。泳動開始位置から先行色素(ブロモフェノールブルー)の距離をRf=1.00 としたときのそれぞれのタンパク質の相対的移動度(表2)を求めます。(表2)で得られた相対的移動度を横軸、分子量の対数を縦軸にとりグラフにプロットして検量線を引きます(図5) 。未知のサンプルの移動度とこの検量線から未知サンプルの分子量を推定します。
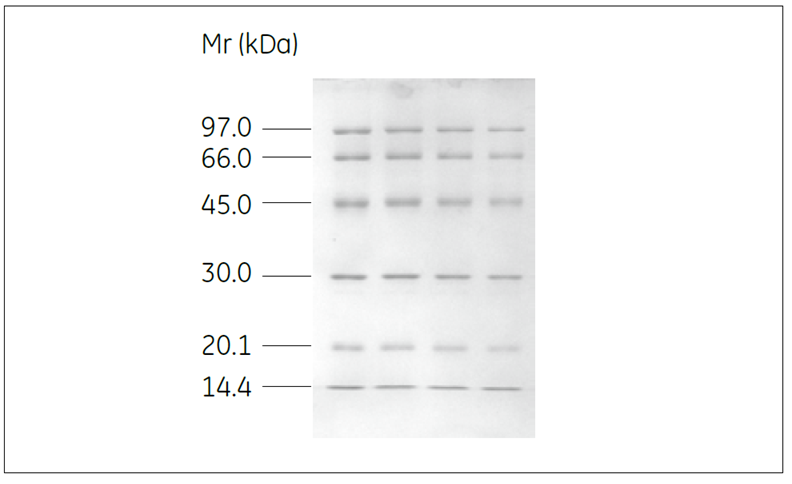
図4.SDS-PAGE のパターン
縦型ミニゲル電気泳動装置を用いてSDS-PAGE を行いました。サンプルバッファーでLMW Marker Kit を等倍希釈して、アクリルアミドゲル(15%T、2.7 %C)にそれぞれ3 μl アプライしました。電気泳動は、20 mA の定電流、55 分間で行いました。泳動後のゲルは、CBB で染色しました。
表2.SDS-PAGE(図4)における各タンパク質の分子量と相対的移動度
| Protein | Mr(Da) | Rf |
|---|---|---|
| Phosphorylase b | 97,000 | 0.07 |
| Albumin | 66,000 | 0.13 |
| Ovalbumin | 45,000 | 0.25 |
| Carbonic anhydrase | 30,000 | 0.46 |
| Trypsin inhibitor | 20,100 | 0.67 |
| α -Lactalbumin | 14,400 | 0.89 |
*分子量の大きなタンパク質はゲルに入りにくい場合があります。
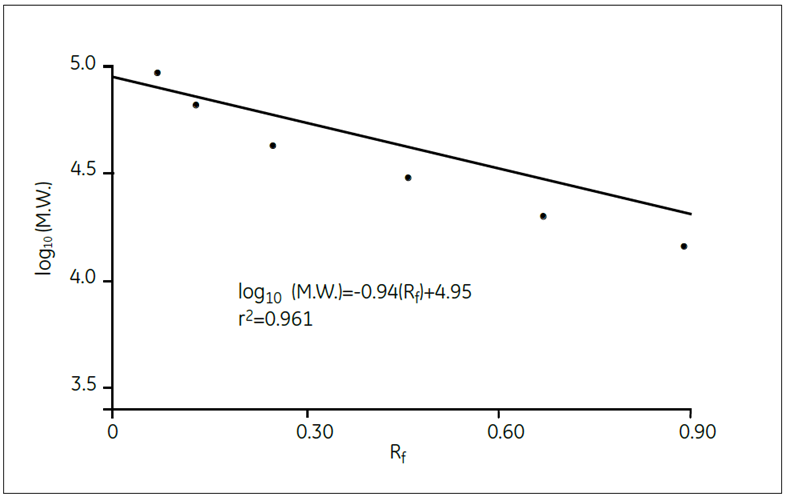
図5.SDS-PAGE による検量曲線
表2 の数値から、最小二乗法で検量曲線の式を求めました。
電気泳動装置の選び方
スクリーニング目的には、泳動距離の短いミニゲル用泳動装置、分離を高め大きく展開するには、泳動距離が長く大きなゲルが泳動できる装置を選択します。さらに、ゲル作製の手間がなく再現性に優れた結果を得るにはプレキャストゲルの使用が有効です。発熱によるバンドの歪みを抑えるために、ゲル全面を冷却できる電気泳動装置を用います。
- スタンダードな装置
SE260
最も一般的なミニゲル(サイズ10 × 10.5 cm もしくは10 × 8cm)を泳動するための装置です。 - ブロッティングもできる一体型装置
miniVE
ミニゲル(サイズ8 × 10 cm)の電気泳動装置およびブロッティング装置としてご使用いただけます。 - 2D とウエスタンブロッティングとの組合せアプリケーションに
SE 600 Ruby / SE 660
ラージゲル(サイズ18 × 16、24 cm)が泳動できる装置です。2D など分離能の高いアプリケーションでお使いいただけます。
※そのほかにも、品質試験などで大量にゲルを扱う方におすすめなプレキャストゲルを用いた全自動電気泳動装置PhastSystem™もご用意しております。
ゲル・バッファーの作製、調整方法
ゲルの作製方法
準備
-
【溶液1:モノマーストック溶液(30.8% T, 2.6% C)】
- Acrylamide PAGE ………………………………… 60.0 g
- N, N'-Methylene-bisacrylamide ………………… 1.6 g
- →超純水で200 ml に調製
- 調製後の溶液は、遮光、4℃で3 ヶ月保存可能です。
- アクリルアミドは神経毒なので取り扱い時には手袋やマスクを着用してください。
-
【溶液2:4 ×分離ゲルバッファー(1.5 M Tris-Cl, pH8.8)】
- Tris …………………………………………………… 36.3 g
- →150 ml の超純水に溶解
- →4N HCl でpH8.8 に合わせた後、超純水で200 ml に調製
-
【溶液3:4 ×濃縮ゲルバッファー(0.5 M Tris-Cl, pH6.8)】
- Tris …………………………………………………… 3.0 g
- →40 ml の超純水に溶解
- →4N HCl でpH6.8 に合わせた後、超純水で50 ml に調製
-
【溶液4:10% Sodium Dodecyl Sulphate(SDS)】
- Sodium Dodecyl Sulphate ……………………… 5.0 g
- →超純水で50 ml に調製
- 室温で保管します。
-
【溶液5:10% 過硫酸アンモニウム(APS)】
- Ammonium Persulphate ………………………… 0.1 g
- →超純水で1 ml に調製
- 用事調製します。
- 超純水を加えたときにパチパチと音がすることをご確認ください。音がしない場合は古い可能性があるため新しいものを使用します。
- n- ブタノールと超純水を1:1 の比でよく攪拌します。上下層に分かれたところで下層の溶液を捨てます。上層の溶液をさらに攪拌して下層の溶液を捨てます。この操作を数回くり返し、上層を使用します。
手順
- 分離ゲル溶液を調製します(表3)。X はアクリルアミド濃度によって変えます(表4)。Y は総溶液量になるように超純水を加えます。
- ガラス板とスペーサー等を用いてゲルサンドイッチを組み立てます。ゲルサンドイッチに(1)の溶液を流し込みます。
- 溶液6 を静かに重層して1 時間静置してゲルを固めます。
- ゲルと溶液6 の境界がはっきりと見えるようになったら、溶液6 を捨て水道水で洗い流します。
- 超純水で表面をリンスして濃縮ゲル(表4)を入れます。
- コームを差込み、そのまま1時間静置してゲルを固めます。
表3.ゲル溶液
| 分離ゲル | 濃縮ゲル | |
|---|---|---|
| モノマーストック溶液(溶液1) | X ml | X ml |
| 4 ×分離ゲルバッファー(溶液2) | 2.50 ml | - |
| 4 ×濃縮ゲルバッファー(溶液3) | - | 1.25 ml |
| 10% SDS(溶液4) | 0.10 ml | 0.05 ml |
| 超純水 | Y ml | Y ml |
| 10% APS(溶液5) | 50 μl | 25 μl |
| TEMED | 5 μl | 2 μl |
| 総溶液量 | 10 ml | 5 ml |
総溶液量はゲルの枚数、ゲル厚に応じて調節します。
APS、TEMED はゲルサンドイッチに流し込む直前に加えてよく混ぜてください。
表4.モノマーストック溶液量(X)一覧
| ゲル濃度(%) | 分離ゲル(ml) | 濃縮ゲル(ml) |
|---|---|---|
| 5 | 1.6 | 0.8 |
| 7.5 | 2.5 | - |
| 10 | 3.3 | - |
| 12.5 | 4.1 | - |
| 15 | 5 | - |
サンプルバッファーの調製方法
サンプル中の塩濃度をなるべく下げ、かつサンプル間の塩濃度差を小さくした後、サンプルバッファーでタンパク質の変性を完全に行います。不完全な変性はアーチファクトの原因になります。通常はサンプルと1. の2 ×サンプルバッファーとを等量混合して使用します。サンプル濃度が低い場合は2. の6 ×サンプルバッファーとを混合して使用します。どちらも泳動前に95℃で3 ~ 4 分間ボイルします。
- 2 ×サンプルバッファー
(0.125 M Tris-Cl pH6.8, 4% SDS, 20% グリセロール, 3.1% Dithiothreitol(DTT))- 4 ×濃縮ゲルバッファー(溶液3) ………………… 2.5 ml
- 10% SDS(溶液4) …………………………………… 4.0 ml
- Glycerol, 85% ………………………………………… 2.4 ml
- DTT ……………………………………………………… 0.31 g
- Bromophenol Blue …………………………………… 0.2 mg
- →超純水で10 ml に調製
- サンプル溶液に等量のサンプルバッファーを加えます。よく混合し95℃で3 ~ 4 分間ボイルして処理します。
- 作製後、1 ml ずつに分注し、‒ 40 ~ ‒ 80℃にて保存可能です。
- 6 ×サンプルバッファー
(0.35 M Tris-Cl pH6.8, 10% SDS, 30% グリセロール, 9.3% DTT)- 4 ×濃縮ゲルバッファー(溶液3) ………………… 7.0 ml
- SDS ……………………………………………………… 1.0 g
- Glycerol, 85% ………………………………………… 3.5 ml
- DTT ……………………………………………………… 0.93 g
- Bromophenol Blue …………………………………… 1.2 mg
- →超純水で10 ml に調製
- サンプル濃度が低い場合に用います。サンプル溶液1 容量に対し、6 ×サンプルバッファーを5 容量加えてよく混合し、95℃で3 ~ 4 分間ボイルします。
- 1 ml ずつ分注し、‒ 40 ~ ‒ 80℃にて保存可能です。
泳動バッファーの調製方法
- 10 ×泳動バッファー(0.25 M Tris, 1.92 M グリシン, 1.0% SDS)
- Tris ……………………………………………………… 30.28 g
- Glycine ………………………………………………… 144.11 g
- SDS ……………………………………………………… 10.0 g
- →超純水で1.0 L に調製
- pH 調整は必要ありません。
- 2 ヶ月間保存可能です。
タンパク質分子量マーカーの選び方と使い方
タンパク質分子量マーカーの選び方
ウェスタンブロッティングに対応した分子量マーカーをご用意しています。下図より目的にあわせたマーカーをお選びください。分子量範囲や検出方法は(表5)よりご覧いただけます。
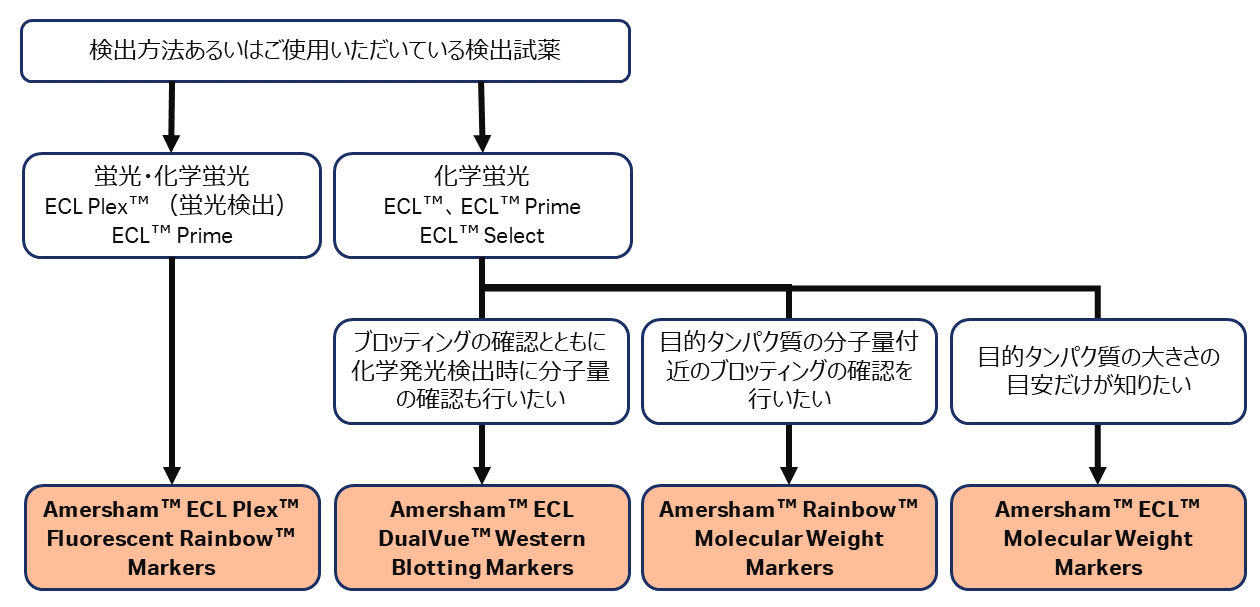
表5.ウェスタンブロッティング用分子量マーカー 一覧
| 製品名 | コード番号 | 検出方法 | 分子量範囲(kDa) | 対応検出試薬 | 包装 | 備考 |
|---|---|---|---|---|---|---|
| Amersham™ ECL Plex™ Fluorescent Rainbow™ Markers | RPN810 | 可視/蛍光 | 12 ~ 225(可視, 蛍光) | ・ECL™ Plex ・ECL™ prime(化学蛍光) |
120 μl(40 ~ 80 回分、ミニゲル時) | Cy™3/Cy™5 による蛍光標識。可視検出も可能。 |
| Amersham™ ECL DualVue™ Western Blotting Markers | RPN810 | 可視/化学発光検出 | 15, 16, 100(可視) 15 ~ 150(HRP) |
・ECL™ ・ECL™ prime ・ECL™ select |
25 回分ミニゲル時 | キット付属のS-Protein-HRPを添加し、化学発光検出。 |
| Amersham™ Rainbow™ Molecular Weight Markers | Full:RPN800E High:RPN756E Low:RPN755E |
可視 | 12 ~ 225(Full,High) 3.5 ~ 38(Low) |
・ECL™ ・ECL™ prime ・ECL™ select |
250 μl(約50 回分ミニゲル時) | 正確な分子量測定には不向 |
| Amersham™ ECL™ Molecular Weight Markers | RPN2107 | 化学発光 | 14.4 ~ 97.0 | ・ECL™ ・ECL™ prime ・ECL™ select |
1 pack(25 回分、ミニゲル時) | HRP 標識ストレプトアビジンを添加し、化学発光検出。 |
ECL Plex™ Fluorescent Rainbow Markers の使い方
蛍光検出可能なマーカーであり、蛍光ウェスタンブロッティング時に利用可能です。レインボータイプの着色マーカーの機能も持ち、可視でも確認が可能です(図6) 。
- 最大検出バンド数:10本
- 分子量(kDa):225 /150 /102 /76 /52 /38 /31 /24 /17 /12
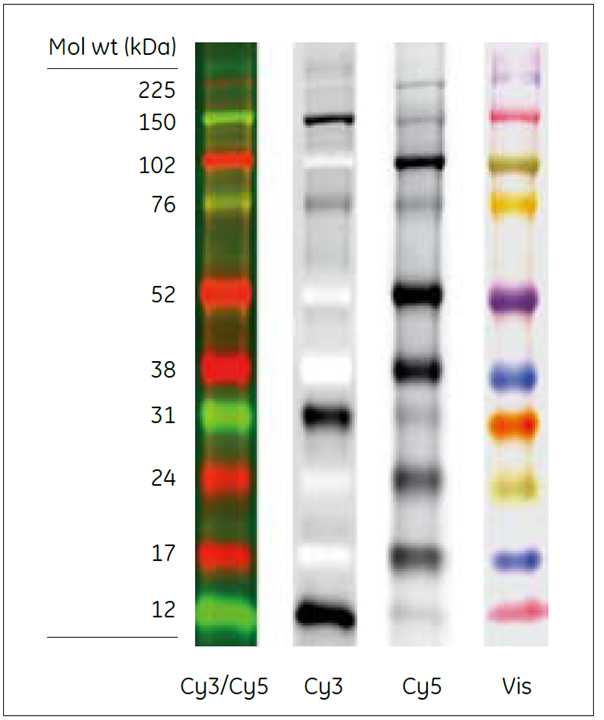
図6.ECL Plex™ マーカー分離パターン
手順
- ECL Plex™ Fluorescent Rainbow Markers を室温で溶かします。
冷凍保存でSDS が沈殿した場合は37℃で短時間温めて沈殿を溶解します。 - ゲルに必要量を添加して電気泳動します。
ゲルサイズによって推奨添加量が異なります。8 × 10 cm のゲルは1.5 ~ 3 μl です。
Cy™2 の検出条件ではバンドは見えません。 - 検出試薬のプロトコールにしたがいブロッティング~検出を行います。
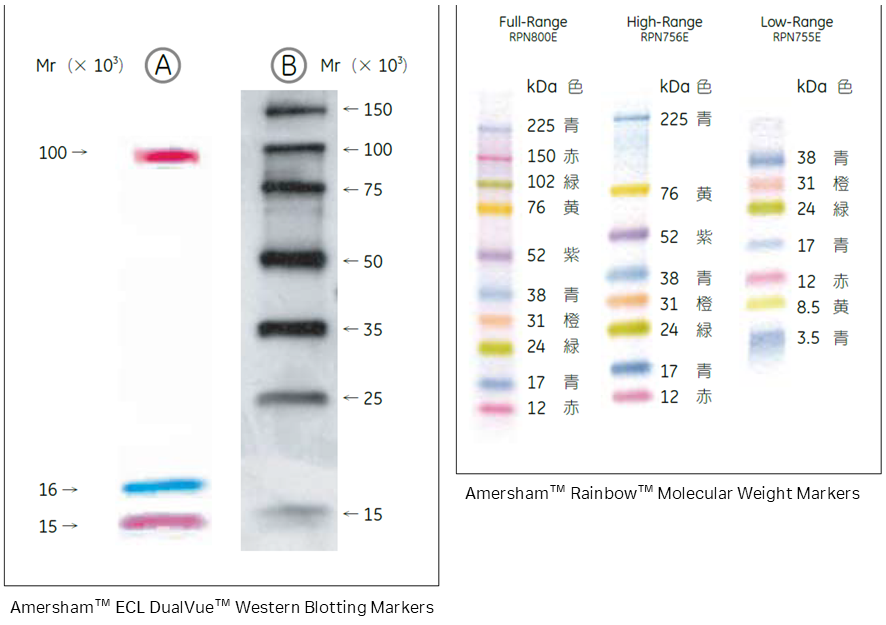
ECL DualVue™ Western Blotting Markers の使い方
電気泳動・ブロッティング状態の目視確認用の3 種類の着色済みタンパク質と、化学発光検出が可能な分子量測定用の7種類のS-tagged 組換えタンパク質がプレミックスされた分子量マーカーです。(図7)抗体反応液に、キット付属のS-Protein-HRP を添加して、マーカーを目的サンプルと同時にECL 化学発光検出することができ、より正確に分子量測定が行えます。
- 最大検出バンド数:3 本(可視)、7 本(化学発光)
- 分子量(kDa):100 /16 /15(可視)
150 /100 /75 /50 /35 /25 /15(化学発光、すべてリコンビナントタンパク質) - 内訳:可視:Phosphorylase b(100 kDa), Myoglobin(16 kDa), Lysozyme(15 kDa)
手順
- 使用直前にECL DualVue™ Western Blotting Markers を室温に戻します。
‒ 15 ~ ‒ 30℃保存により、マーカー溶液中のSDS が沈殿した場合は、37℃で数分温めて沈殿を溶解させます。 - マーカー溶液をよく混合し、マーカー溶液5 μl に対して、2 ×サンプルバッファーを5 μl 加えて混合します。
- 溶液をゲルのウェルあたり10 μl 添加して泳動します。
- 電気泳動後、タンパク質をメンブレンにブロッティングします。
ブロッティングの条件は、ご使用のブロッティング装置のマニュアル等に従ってください。
転写後、着色済みマーカーがメンブレン上に目視で確認できます。 - ブロッキングと一次抗体反応を、サンプルに適した条件で行います。
- HRP 標識二次抗体の反応液にキット付属のS-Protein-HRP conjugate を、ECL™ 検出試薬の種類にあわせて(表6)の希釈倍率になるよう加えます。
ECL DualVue™ Western Blotting Markers はS-tag に対するS-Protein の特異的な結合を利用しています。目的サンプルがS-tagged 組換えタンパク質の場合は、交差反応が起こりますので、分子量マーカーのレーンだけを切り離し、別々のイムノディテクション操作を行ってください。最後にECL™ 検出試薬との反応後、切り離したものの位置をあわせてフィルムの露光を行ってください。S-Protein とS-tag の結合の原理については以下の文献をご参照ください。
Richards,F.M and Wyckoff, H.W. The Enzymes, Vol. IV (Boyer, P.D. Ed) p.647-806, (1971) Academic Press, New York. Kim,J. S. and Raines, R.T. Protein Sci. 2, 348-356 (1993) - 室温で30 分以上インキュベートします。
- 以後、化学発光検出試薬のプロトコールにしたがい検出を行います。
X線フィルムでの検出の場合、露光時間は1 ~ 2.5 分での検出をおすすめします。
表6.S-Protein HRP conjugate の希釈条件(X 線フィルムによる検出)
| 化学発光検出試薬 | Hybond-ECL / ニトロセルロースメンブレン使用 | Hybond-P/PVDF メンブレン使用 |
|---|---|---|
| ECL™ | 1:5,000 | 1:10,000 |
| ECL™ Plus | 1:10,000 | 1:20,000 |
| ECL Advance™ | 1:100,000 | 1:200,000 |
CCD イメージャーで検出する場合は、感度にあわせてS-Protein HRP conjugate の希釈濃度を検討します。X 線フィルムの場合の10 倍濃度を目安に検討してください。
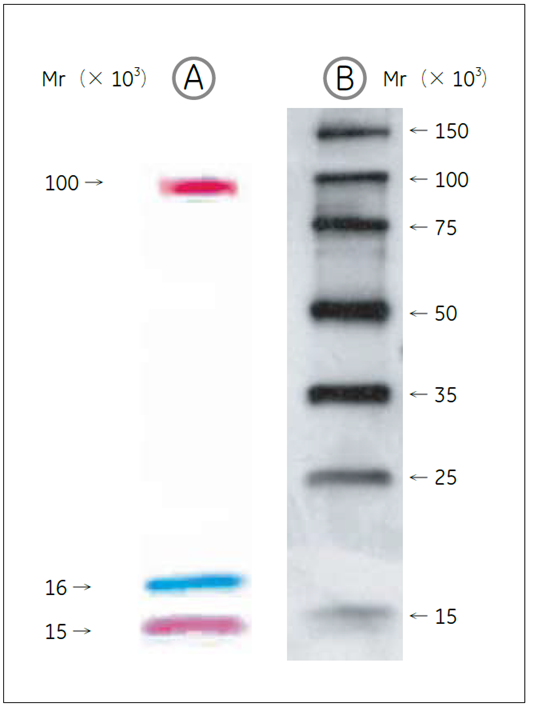
図7.ECL DualVue™ Western Blotting Markers の分離パターン
A:4 ~ 20 % ゲルでSDS-PAGE 後、Hybond-ECL ニトロセルロースメンブレンにブロッティングした状態。3 本のシャープな着色バンドが目視確認できます。
B:AのブロットをS-Protein-HRP とECL™ を用いて検出した結果(Hyperfilm ECL™ に1 分間露光)。7 本のS-tagged タンパク質のバンドが検出できます。
Rainbow™ Molecular Weight Markers の使い方
各バンドが異なる色素で着色されている分子量マーカーです。電気泳動経過や、ブロッティ ング時のトランスファー効率を視覚的に確認できます(図8) 。
Rainbow™ Molecular Weight Markers は、マーカータンパク質に色素が吸着しているため、正確な分子量測定には向きません。分子量の目安としてご使用ください。

図8.Rainbow™ Molecular Weight Markers の電気泳動図
手順
- Rainbow™ Molecular Weight Markers を室温で溶かします。
冷凍保存でSDS が沈殿した場合は37℃で短時間温めて沈殿を溶解します。 - ゲルに必要量を添加して電気泳動します。
ゲルサイズによって推奨添加量が異なります。8 × 10 cm のゲルは5 μl、20 × 20 cm のゲルは10 μl です。 - 検出試薬のプロトコールにしたがいブロッティング~検出を行います。
SE 260 での電気泳動方法
標準的な電気泳動装置の手順をご紹介します。
ゲルサンドイッチの準備
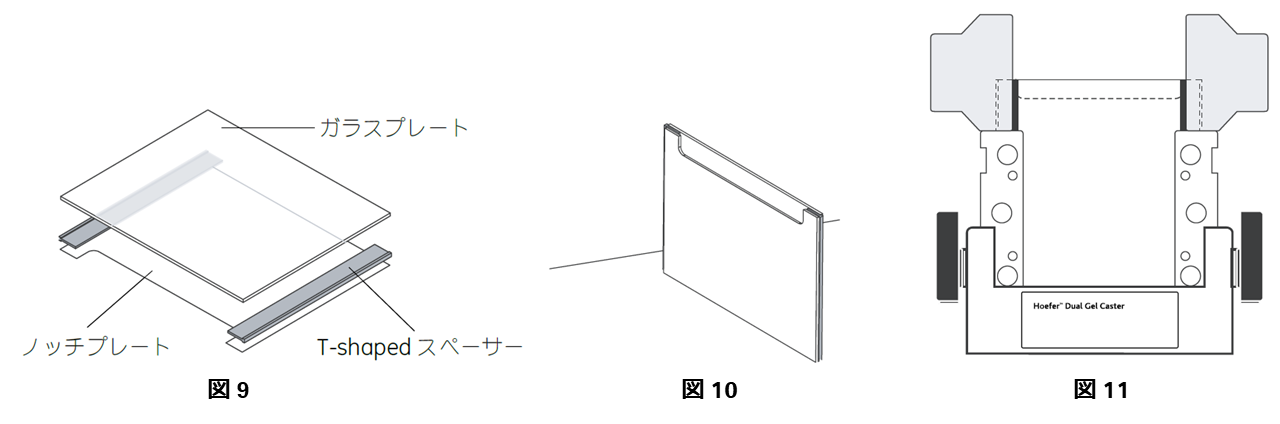
- ゲル枚を固定するための装置、ゲルキャスター(図11)を分解し、水で洗浄します。ガラス板、スペーサーなどもよく洗浄し乾かしておきます。
- ガラスプレートとノッチプレート、T-shaped スペーサーを(図9)のように組み立てます。
- ガラスプレートを立てて、ガラスプレートとスペーサーの下端があうように調整します(図10) 。
- ゲルキャスターに設置します(図11) 。
- ゲル溶液を準備します(枚数、厚さによって溶液量が異なります)。
- 10% 過硫酸アンモニウム、TEMED を加えた分離ゲル溶液をゲルサンドイッチへ流し込み、n-ブタノール飽和水溶液を重層して1 時間以上重合させます。
- n-ブタノール飽和水溶液を除き、濃縮バッファーで分離ゲル表面を洗います。
- 濃縮ゲル溶液を重層して静かにコームを差し込み、1 時間以上重合させます。
泳動槽の準備
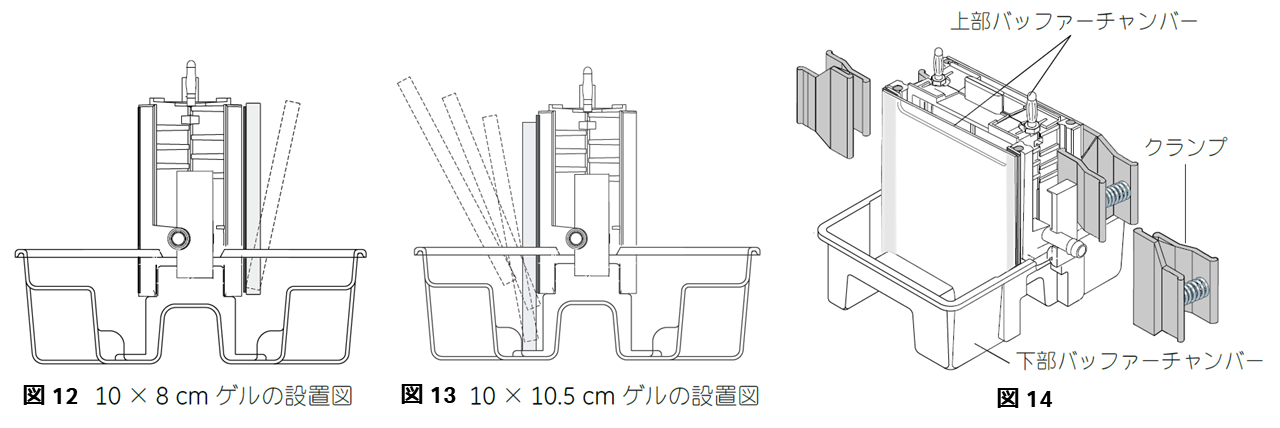
- ゲルサンドイッチを設置します(図12、13) 。ゲルサイズによって設置する位置が異なります。
- クランプで泳動槽とサンドイッチを固定します(図14 )。
サンプル準備
- サンプルを準備します。サンプルの量は下記の表を参考にします。
表7.添加するサンプル量
ウェル数 ゲル厚 0.75(mm) 1.0(mm) 1.5(mm) 5 9.5 μl 12.7 μl 19.1 μl 9 ー 5.8 μl ー 10 3.6 μl 4.8 μl 7.2 μl 15 2.2 μl 2.9 μl 4.4 μl 18 ー 2.9 μl ー 泳動
- 上部バッファーチャンバーに泳動バッファーを入れます。1 チャンバーあたりおよそ75 ml のバッファーが入ります。
- 下部バッファーチャンバーに泳動バッファーを入れます。1 チャンバーあたりおよそ250 ml のバッファーが入ります。電極がバッファーに浸かっていることを確認します。
- シリンジでウェルを洗い、サンプルを添加します。
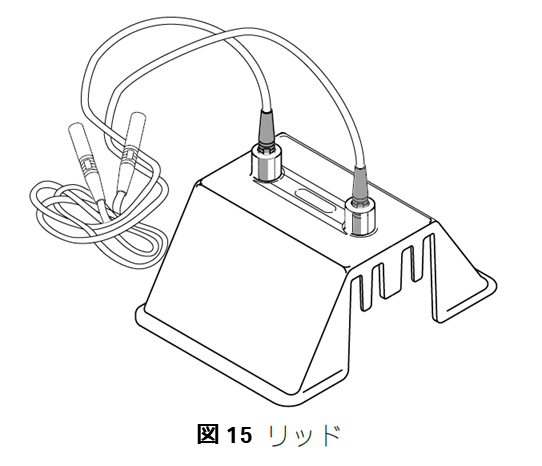
- リッド(図15)を取りつけて定電流もしくは定電圧で泳動をはじめます。ゲルあたり20 mAで泳動します。2 枚泳動する場合は40 mA になります。
- 色素がゲル下端に達する直前に泳動を終了します。
泳動終了
- 電源を切り、リッドをはずします。
- 泳動バッファーを捨て、ゲルサンドイッチを取りはずします。
- プレートからゲルをはがし、ブロッティングもしくは染色を行います。ブロッティングを行う場合、ゲルをブロッティングバッファーに浸して平衡化します。
ゲル染色試薬の選び方
電気泳動でタンパク質がうまく分離されていることやブロッティング後にタンパク質のブロッティング効率を確認するためにゲルを染色してバンドを可視化することができます。一般的にはCBB 染色が用いられます。CBB とDeep Purple はメンブレンを染めることもできます。お持ちの装置やサンプル量から手法をお選びください(下フロー図)。また、各手法の仕様を表8 に示しました。
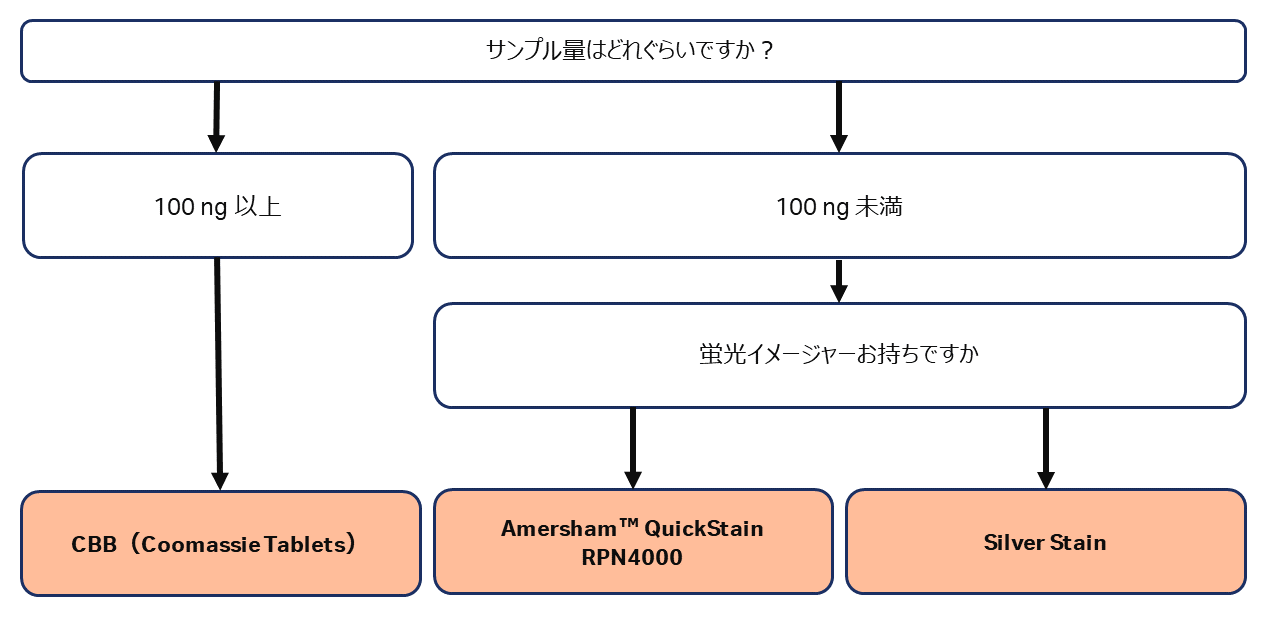
表8.染色試薬の仕様
感度 定量性 操作性 検出所要時間 タンパク質の修飾 CBB 5 ~ 100 ng ◯ 簡便 3 時間~終夜 なし Silver Stain 0.3 ~ 0.5 ng △ 煩雑 ~ 4 時間 あり Amersham™ QuickStain <数ng ~ ◎ 簡便 ~ 4 時間 なし 関連記事
ウェスタンブロッティングとは?
ウェスタンブロッティングは、電気泳動の優れた分離能と抗原抗体反応の高い特異性を組み合せて、タンパク質混合物 から特定のタンパク質を検出する手法です。タンパク質の存在を検出するだけでなくタンパク質の状態確認(リン酸化などの修飾)もできます。リン酸化を介したシグナル伝達機構の解析や狂牛病の二次検査での異常型プリオンの検出などライフサイエンスの様々な分野で、目的タンパク質の検出や解析に利用されています。
ブロッティングとは?
電気泳動後のゲルからメンブレンにタンパク質を転写(ブロッティング)する方法をご紹介します。
抗体反応と検出とは?
ブロッティング後のブロッキングおよび一次・二次抗体を用いた反応についてご説明します。メンブレンから抗体を除去して別の抗体を使って検出する方法(リプロービング・抗体除去)もご紹介します。
ウェスタンブロッティング:トラブルシューティング
実際のトラブル写真を掲載して解決策や改善のためのワンポイントをご紹介します。
ウェスタンブロッティング攻略ガイド抜粋版PDF
ウェスタンブロッティングで初めて化学発光検出の実験をされる方にも使っていただけるよう、標準プロトコールをご用意しました。
関連製品
Amersham™ ImageQuant™ 800
富士フイルムと共同開発したCCDイメージャーの最新機種。いままでよりもっと簡単にきれいな画像を撮影したい方におすすめです!
Amersham™ Typhoon™ scanner
Typhoon™ FLA 9500シリーズの後継機種で富士フイルム社と共同開発したスキャナータイプ画像解析装置
Amersham™ ECL™ シリーズ
ウェスタンブロッティングに適した化学発光検出試薬、Amersham™ ECL™シリーズです。
Amersham™ QuickStain
Cy5とラベリングバッファーのキットで、SDS-PAGEのバンドの蛍光検出やウェスタンブロッティングのメンブレン上で抗体検出バンドとトータルプロテインの蛍光検出を同時に行うことによりノーマライズすることがが可能となります。
お問合せフォーム
※日本ポールの他事業部取扱い製品(例: 食品・飲料、半導体、化学/石油/ガス )はこちらより各事業部へお問い合わせください。