DIGE 道場 第2回
こんなにすばらしい!2D-DIGE法
第2回 もくじ
- 二次元電気泳動に対する誤解と2D-DIGE
- 2D-DIGEの魅力
- 2D-DIGEが可能にした「レーザーマイクロダイセクションからのプロテオーム解析」
- 最後に
Dr. 近藤のコラム
 コラム第2回 DIGEなおサイフの話 - 2D-DIGEは本当に高い?
コラム第2回 DIGEなおサイフの話 - 2D-DIGEは本当に高い?
2. 2D-DIGEの魅力
2-1 再現性の大幅な向上
●二次元電気泳動法の再現性を左右する3つの要素
古典的な二次元電気泳動法では、一枚のゲルごとに一種類のサンプルを泳動するのが原則である(注1)。そこで問題になるのがゲル間で発生する泳動パターンのばらつきである。すなわち「同一のチューブからとったサンプルを2枚のゲルに泳動したときに、ゲル間で泳動パターンが異なる」ということである。この現象をもってして「二次元電気泳動は再現性が悪い」と結論される(注2)。
この問題には三つの要素が含まれている。
- サンプルの不均一性 -本当に “同じ” サンプルを泳動できているのか?-
- 電気泳動操作から生まれるばらつき
- サンプル添加量
これらについて、ひとつずつ詳細を見てみたい。
1. サンプルの不均一性 -本当に “同じ” サンプルを泳動できているのか?-
一つ目は、同じサンプルを2枚のゲルに泳動していない可能性がある、ということである。同じチューブからサンプルをとったからといって、同じ内容のタンパク質をとっているとは限らない。DNAが多量に含まれる粘度の高いサンプルの場合、チューブのどこからサンプルをとるかによって、実際にゲルに添加されるサンプル状態は変わっている可能性がある。サンプルを凍結保存した場合には、その前後でタンパク質の可溶化の度合いが変わっている可能性もあるだろう。
すなわち、二次元電気泳動法の再現性の問題ではなく、サンプルの質の問題、サンプルの調製法に問題があると言える。
2. 電気泳動手技から生まれるばらつき
二つ目は電気泳動自体のばらつきである。サンプルの添加方法、一次元目等電点電気泳動のゲル(IPGゲル)のSDS平衡化処理と二次元目SDS電気泳動ゲルへの添加、SDS-PAGEゲル作製、温度コントロール、などがばらつきの原因となりうる。厳守するべきポイントが強調されていないプロトコールを用いた場合にこれらの問題が発生する可能性がある。詳しいことは次号(第3回)以降に記載するが、IPGゲルの平衡化処理を例に挙げて紹介したい。
IPGゲルの平衡化処理は室温で行うことになっている。しかし、この条件を守るためには、通常の実験室で実験を行っていればいいというものではない。平衡化バッファーにはウレアが含まれているので、調製した時点では必ず溶液の温度は室温以下である(ウレアの吸熱反応のために)。冷たい平衡化バッファーを使って処理をした場合には室温で実験をしていることにならない。そして冷たい平衡化バッファーを使用すると、かなり高い確率で泳動に失敗する。同様に、等電点電気泳動後に?80℃で保存していたIPGゲルを使う場合に、フリーザーから出してすぐの凍ったままのものを使うと、同じ理由から実験に失敗する可能性が高い。このような場合に、筆者が目にしてきた泳動パターンでは、どうもSDS化がうまくいっていないケースが多いように感じる。
筆者のラボでは朝一番にDTT以外を含む平衡化バッファーを作製し、室温で昼過ぎまで撹拌し、十分室温になってから使用することにしているし、IPGゲルはしばらく室温に放置し完全に溶けてから使用している。特別なことをしているのではなくプロトコールに忠実に実験をしているだけなのだが、ここまで気を配っていないラボは多いのではないだろうか?
3. サンプル添加量
三つ目はサンプルの添加量である。電気泳動に添加できるタンパク質のキャパシティーについて、再現性よくパターンが得られる量と、泳動パターンがどうであろうと、とにかく添加できる限界の量とが区別されずに記載されることがある。
特定のタンパク質、不要物、またはサンプル中に混在する泳動阻害物質の含有量が増えるなどの要因から、一般には多量のタンパク質を添加するほど再現性は劣化しがちである。境界領域で実験をしていれば、なおさら再現性は悪くなることが推測できる。塩などの泳動を阻害する物質についても同様のことが言える。「再現性よく泳動できる塩濃度」と「泳動可能な塩濃度」は異なるのである。
●誰でも実践できる、3つの要素の解決法とは?
上で挙げたどの要素も、条件が適切に整えられた実験環境では解決可能である。
例えば、1番目のサンプルの不均一性の問題は、粘度を与える夾雑物の少ない検体が得られるプロトコールを用いることで解決可能である。血清タンパク質の二次元電気泳動パターンは異なる研究グループ間でもかなり一致することが以前から知られている。これは泳動を阻害する夾雑物の混入を少なくすることが簡単だからだろう。また、凍結融解後のサンプルはしっかり溶かすか、不溶物を除去することが必要である。
2番目の実験手技の問題は、ポイントとなる操作ステップを理解した上で、きちんとプロトコールに記載して徹底しさえすれば解決可能である。二次元電気泳動法は工数が多いので、重要な点が指摘されていないプロトコールでは失敗する可能性も高くなる。これらのポイントについては、この連載にたっぷりと盛り込んでいくので、参照されたい。
3番目のタンパク質添加量の問題も、解析ゲルと分取ゲル(タンパク質同定用にスポットを切り出すためのゲル)を分けて泳動すれば問題ない。解析用のゲルから直接スポットを切り出そうとすると、どうしても多量のタンパク質を添加したくなるものだが、筆者の考えるプロトコールでは、分取用ゲルには100 µgのタンパク質を添加すれば十分と考えている。
そうは言ってもプロテオーム解析に供される検体はさまざまであり、実験も一年中息を詰めるように集中できるわけではない。一定の幅をもってどんな検体に対してもルーチンに機械的にかつ臨機応変に対応するにはある程度の経験が必要だった(注3)。これはどの実験についても言えることで、二次元電気泳動法だけ例外な訳ではない。
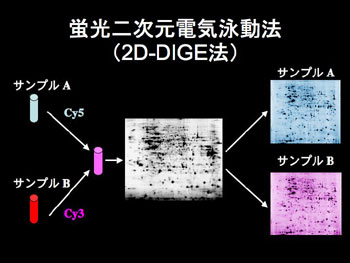 図1 2D-DIGE法の最も基本的なアプリケーション
図1 2D-DIGE法の最も基本的なアプリケーション
2つのサンプル(サンプルAとB)をそれぞれ異なる蛍光色素(Cy5とCy3)で標識する。両者を混合して一枚のゲルで二次元電気泳動法を行う。
●2D-DIGE法による再現性の向上 -内部標準サンプルの利用-
2D-DIGE法では複数のサンプルを異なる蛍光色素で標識し混合してから一枚のゲルで泳動し分離する(図1)。したがって、一枚のゲルで複数のタンパク質サンプルの比較が可能になる。ゲルの泳動パターンに左右されることなく同一のゲル内では比較解析ができる。
しかし、この実験デザインは実際の実験を考えるとあまり現実的ではない。蛍光色素の種類の数しか一枚のゲルに泳動できないからである。臨床検体を用いた実験では100検体くらいのプロテオーム解析は必須なのだが、100種類の蛍光色素をつくることはできないだろう。仮にできたとしても、100種類のサンプルを一枚のゲルで泳動することは、ゲルに添加できるタンパク質量の限界を考えると不可能である。
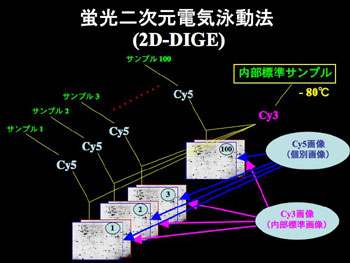 図2 多数検体のための2D-DIGE法解析フロー
図2 多数検体のための2D-DIGE法解析フロー
蛍光色素の数を超えるような多数のサンプルを扱う場合、2D-DIGE法では、「内部標準をとる」プロトコールを使う(図2)*。筆者のラボでもルーチンに行われている方法である。
具体的には、調べたいすべてのサンプルから等量ずつタンパク質を集めて混ぜ合わせ、それを内部標準として分注し保存する。実験ごとにこの内部標準サンプルを取り出してCy3で標識し、個別のサンプルはCy5で標識する。このようにして異なる蛍光色素で標識されたサンプルを混合し一枚のゲルで泳動し分離する。図2では100検体を泳動するプロトコールを模式化した。泳動後のゲルを蛍光スキャナーのCy3の波長でスキャンすれば内部標準サンプルの画像、Cy5の波長でスキャンすれば各検体サンプルの画像を得ることができる。
内部標準を使った解析手法の妙味は、
「内部標準となるサンプルに、すべての検体サンプルに含まれるすべてのタンパク質が、平均値のボリュームで含まれている」
ということである。各検体に含まれるすべてのタンパク質スポットを有した “同じサンプル(=内部標準)” を “異なるゲル” に泳動することで、ゲル間のばらつきを補正することができる。また、ゲル内では、各検体の泳動パターン(Cy5)を内部標準の泳動パターン(Cy3)で補正することで、各スポットを定量的に扱うことができるようになる。しかも、この補正は、2D-DIGE法に対応した画像解析ソフト(DeCyder™ 2D Software (Cytiva)やProgenesis SameSpots (Nonlinear Dynamics社))が自動的に行う。
ゲル間の補正が可能といっても、ゲル間のパターンがあまりにも違うと補正しきれない。Cy3画像上(内部標準の泳動パターン)でコンスタントに観察されるタンパク質スポットのみが解析対象である。つまり電気泳動の実験手技的に再現性の高いスポットが解析の対象となる。
また、この方法では上述の1番目の問題点である同一サンプルの不均一性は解決されない。したがって、筆者のラボでは混合後のサンプルを3等分して3枚のゲルで泳動し、3枚のゲルのデータの平均をとることにしている。図2のように100種類のサンプルを解析する場合、実際には300枚のゲルを泳動している。300枚のゲルと言うとちょっと引いてしまう方もおられるかもしれないが、後述するように2D-DIGE法ではスループット性よく実験ができるので、研究環境さえ整えれば300枚くらいの泳動はまったく問題ではない。
次へ 2-2. 定量性の大幅な向上
注釈
注1:応用技術として、アイソトープ標識していないサンプルをアイソトープ標識した微量サンプルと混ぜて泳動し、まず銀染色で前者のタンパク質スポットを観察できるようにし、次に同じゲルを乾燥させてX線フィルムに感光することで後者のタンパク質スポットを検出する、ということはされていた。
注2:実験系の再現性を評価することは難しい。二次元電気泳動法自体の再現性を論じるのであれば、精製された単品のタンパク質をくり返し泳動することになるのだが、それは現実的な状況を再現していることにならない。そこで細胞や組織から抽出したサンプルで実験が行われることになる。そうするとサンプリングの再現性の問題が浮上してくる。サンプリングの再現性は二次元電気泳動法に固有の問題ではないが、実際には解決しなくてはいけない問題である。
注3:「ある程度の経験」の定義が難しいのだが、学生時代からの長年のトレーニングが必要だとか特殊な才能や筋肉や神経が必要というわけではない。一般的な実験並みに指導とトレーニングが必要、という程度である。
参考文献
- Kondo T, Hirohashi S. Nat Protoc. 1(6):2940-56(2006)

